How to guide: Assessing, arranging and confirming capability and capacity
Capacity and capability (C&C) assessment is the method by which sites consider whether they can take on a new study. In some cases this needs to be a formal assessment and confirmation, in other cases an email may suffice, or in others the Health Research Authority (HRA) may state that the study can proceed without C&C unless the site indicates any objection. Regarding C&C for Participant Identification Centre (PIC) activity, please see the ‘Understanding the role of PICs’ guide, and the below PIC section.
This guide outlines the ways in which practices should undertake the appropriate Assess, Arrange and Confirm Capacity and Capability stage of site agreement when setting up a study.
See also HRA guidance on: Assess, arrange and confirm terminology.
Key information you should be aware of:
-
West Yorkshire Research & Development (WY R&D) issues an advisory email for studies of which we have had sight, to assist practices with completing their review. Practices can request a copy of this advisory from WY R&D or from the study team.
-
Research governance is conducted centrally by the HRA. The HRA issues HRA approval which incorporates ethical review.
-
Each participating site must assess, arrange and confirm their capability and capacity to participate, dependent on the requirements of the individual study.
-
The decision to participate, and the agreement, should be at the level of the participating organisation, i.e. usually at the level of the practice (as an independent contractor). PCNs are not able to sign a contract unless they have been set up as a company, and neither a PCN nor a federation is not able to sign a contract on behalf of its members unless there is a formal agreement that they can do this. If the research is taking place at the federation level, e.g. in the out of hours service, then this could be signed off by the federation. The ICB cannot sign off on behalf of practices.
-
It is important to note that the processes confirmed in the HRA approval letter must be followed as formal confirmation or signing of agreements is legally necessary where indicated in this letter.
Highlights
-
What can happen before approval
-
Process following approval
-
How to assess
-
How to arrange
-
How to confirm
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Principal Investigator guide
-
Data guide
Full guide
Pre-HRA approval:
-
Sponsors are encouraged to liaise with potential sites as early as possible in the process of developing their study. This may be before HRA approval for the study has been received or even applied for.
-
At the pre-approval stage, sites can carry out a feasibility review of the study documents, but should be aware that the documents cannot be considered final until HRA approval is confirmed to the study team.
-
A patient could be involved to discuss study feasibility, but no patient recruitment can usually take place until the study is approved.
After HRA approval of the study:
-
The next step in the process is that the study team will receive a HRA approval letter.
-
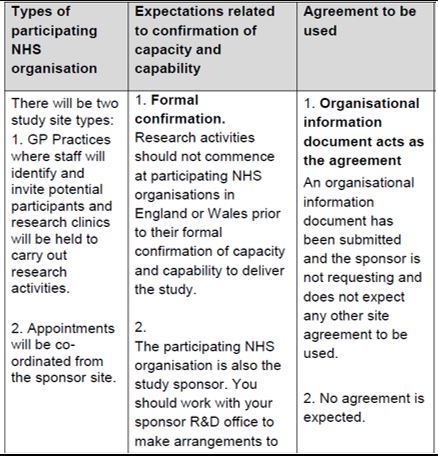
When the HRA approval letter is received, it will confirm expectations related to confirmation of capacity and capability (in the ‘Information to support study setup’ table at the end of the letter).
-
This will include stating whether formal confirmation of C&C (capability and capacity) is required, and whether the study intends to use the Organisation Information Document (OID) as the site agreement.
-
You should ask for a copy of the letter and check this table first to inform what you need to do next.
-
WY R&D should also have the opportunity to issue an advisory email which will give advice on next steps in assessing, arranging and confirming C&C.
-
If formal confirmation of C&C is required, see below for detail of the steps.
Assess:
Further to any initial feasibility assessment done at the Expression of Interest (EOI) stage, this is the main and formal feasibility assessment. When formally confirming your capacity & capability, and/or confirming the OID or contract, this is where you commit to the study, so this assessment should be more in-depth. Depending on the study commitment, this may be quick (for example if just a staff survey), or may require more consideration.
At this stage, you should have from the study team:
-
A copy of the study protocol and local documents (the Local Information Pack).
-
A copy of the Organisation Information Document or study contract.
An advisory email will have been sent to the study team from WY R&D, and you can request a copy of this from the study team or from WY R&D.
At the 'assess' stage you should review the study protocol and/or OID/contract and consider the following:
|
Area |
Things to consider |
Specific questions to ask |
|
Suitable Investigator & Site Personal identified The Ready for Review and HRA Approval letters will confirm if a Principal Investigator (PI), a Local Collaborator or neither is required at participating organisations. Where a PI is required, the participating organisation has identified a suitable investigator. |
‘Suitable’ defined as a named investigator taking into account his/her professional qualification, knowledge of research field, expertise in the procedures involved, previous research experience, training in research methods (including informed consent), training in Good Clinical Practice (if applicable) and ability to take clinical responsibility for local research team. |
Do you have suitable staff who can give enough time to the study; can they be released to work on the study? Are they engaged and interested in the research? Do they have the required skills? Has a suitable and willing PI been appointed (from the practice team?) |
|
Conflict of Interest |
Any personal involvement of site personnel with the sponsor or funder have been declared |
Do any of the staff have any conflicts e.g. with other roles, personal relationships? |
|
Study Follow-up |
The clinical research staff or participating organisation have the capacity to support all study follow up as required |
Are you aware of the extent of follow-up activity – will there be ongoing commitments after recruitment? (e.g. follow up appointments, data from the clinical system) |
|
GCP Training |
All staff that require GCP training have accredited/appropriate training eg NIHR or similar standard |
Have the staff had GCP or other required training? |
|
Appropriate Equipment available |
Study specific equipment is on site or being provided by sponsor |
Does the study require any specific equipment and does the practice have this available? |
|
Safety |
The requirements for reporting to the sponsor on progress and pharmacovigilance |
Do you understand what reporting is required? |
|
Local arrangements for handling of Investigational Medicinal Product (IMP) |
Are there specific considerations for handling the Investigational Medicinal Product? |
|
|
Local Informed consent needs |
Obtaining consent in accordance with the study protocol while considering the needs of participants who may not adequately understand verbal explanations or information written in English |
Are there any concerns about your population engaging with the informed consent process, e.g. language needs. Any translated versions required are the responsibility of the study team. |
|
Training |
The participating organisation can accommodate training format required by the Sponsor e.g. online, face to face or routine briefings within the specified time frame required by the Sponsor |
Do you know what site initiation/training will be provided by the study team and are you able to accommodate this? (e.g. consider timing, face to face vs virtual). |
|
HR |
Local Human resource requirements to ensure that staff only undertake study specific activities that are appropriate to the job and competencies of the individual, and that appropriate supervision will be provided as required. This may include arrangements to issue letters of access or an Honorary contract where participants will be seen by non-NHS Organisation employees |
Have you reviewed the study and ensured that the staff working on the study meet the study requirements (e.g. qualified doctor, nurse, prescriber)? Are there any external staff involved in the study at the practice – these should be referred to West Yorkshire R&D for a letter of access. |
|
Recruitment Target |
Based on the projected study timelines there are sufficient numbers of potential participants meeting the inclusion/exclusion criteria when taking into account any competing trials currently open or planned to open at the participating organisation to minimise the potential for inadequate patient recruitment or non-completion of research |
How many participants does the study wish to recruit from the practice? Do you have enough eligible patients (bearing in mind that you will not manage to get every eligible patient to agree to take part)? |
|
Suitability of site |
Suitability of participating organisation facilities including; adequacy of facilities for any novel procedures or for procedures not part of existing clinical activity; availability and access to resources, facilities, equipment and storage considering impact on current levels of use for non-research activities; and considering Sponsor quality expectations. Availability of work space for sponsor team monitoring if required |
If being asked to undertake activities which are not part of standard clinical practice, are your rooms/facilities/equipment suitable? Do you have enough space/equipment so that taking part in the research will not impact on delivery of standard care? |
|
Indemnity |
The HRA ensures that the sponsor has appropriate insurance for the design and management of the research. It is then a sponsor responsibility to assure itself that each individual site has appropriate insurance for the delivery of the research. For NHS sites, this is inherent and derived from NHS Litigation Authority. For non NHS sites and independent contractors, acting as a site, they will need to arrange suitable indemnity themselves, which should be reviewed and deemed appropriate by the sponsor. |
General practices’ delivery of research is now covered by CNSGP (https://www.hra.nhs.uk/planning-and-improving-research/best-practice/nhs-site-set-up-in-england/frequently-asked-questions-about-research/) |
Guidance [first two columns] provided by the NIHR Clinical Research Network (now Research Delivery Network: RDN) (https://www.rdforum.nhs.uk/content/wp-content/uploads/2015/10/UTF-8CRN-principles-for-local-capacity-and-capa.pdf)
Arrange:
At this stage, the practice should put any necessary arrangements in place to deliver the study. Even when studies don’t require formal confirmation of C&C, there may be some arrangement required.
Please note that, if the study requires software to be added to the practice computers, this usually requires involvement from IT and you can seek advice from WY R&D.
The practice should consider each of the following arrangements, although these will vary by study:
|
Financial arrangements |
The financial arrangements applicable to the participating organisation are adequately described, i.e. the practice has full information/confirmation about which organisations will be paying for which costs. Any costs that the practice incurs should be covered by one of the following:
|
|
Internal arrangements |
Suitable arrangements for permissions/peripheral clinics/access when study involves NHS patients external to the participating organisation (e.g. another NHS Organisation) |
|
Emergency procedures |
Emergency procedures that may be necessary are conducted at the participating organisation in accordance with the protocol (e.g. to protect the participant in the event of a life-threatening incident or adverse event) |
|
Local contact information |
Where applicable the participant is clear about the local address and telephone number (usually included in the patient information), additionally contact details of the local investigator(s), and if applicable, other members of the research team, e.g. research nurses, Emergency contact information, if appropriate, contact information for complaints and, where appropriate, independent advisors. |
|
Archiving and storage arrangements |
Storage of data during the study and any post study archiving arrangements |
|
HRA guidance |
The HRA Approved protocol and any guidance provided in the HRA Approval letter are adhered to. This includes, but is not limited to, any patient consent, Information Governance, Human Tissue Act, and Mental Capacity Act arrangements. Radiation The Ionising Radiation (Medical Exposure) Regulations and the Medicines (Administration of Radioactive Substances) Regulations |
|
Fraud and Misconduct |
Local systems in place for dealing with fraud and misconduct |
Guidance provided by the NIHR Clinical Research Network (now Research Delivery Network: RDN) (https://www.rdforum.nhs.uk/content/wp-content/uploads/2015/10/UTF-8CRN-principles-for-local-capacity-and-capa.pdf)
Confirm:
At this stage, the practice confirms its participation (and confirms they are ready to start). The ways of doing this are varied depending on the study – this is confirmed in the HRA approval letter.
|
Involvement |
HRA letter wording |
What the practice must do |
|
PIC |
|
The HRA letter details the type of agreement the study is using. For a PIC, the HRA recommends that the model PIC agreement is used. This agreement is made between the practice and the site where the research activity is taking place. |
|
No formal confirmation required |
|
This is usually only for studies with minimal 'ask' for practices, or urgent public health studies where it is expected that all practices will participate. The research can proceed without agreement from the practice, after the length of time detailed in the HRA letter (in this example seven days). It is subject to the following conditions (as well as any others detailed in the letter):
If a practice does not wish to participate, they must give the sponsor a reason. If a practice positively confirms they can participate, then the study can proceed earlier than the stated length of time. Guidance can be read here: Where no C&C is required. |
|
Practices required to formally assess, arrange and confirm their participation |
|
The letter should also detail the method by which the practice will confirm C&C. Formal written confirmation must happen before any research activity can take place. This doesn’t mean wet ink signed contracts – in many cases a simple email will be acceptable as confirmation.
|



The next column in the table indicates the agreement to be used to confirm participation. See also the guide for contracts. Although the OID or the model agreement are the preferred contracts, you will need to use the contract which has been submitted and agreed by the HRA.
Most commonly, the standard agreements are used:
|
|
An authorised member of staff from the practice should check through the Localised Organisation Information Document (OID). This should detail what is required by the study, and all of the fields marked with a * should be completed by the study team, outlining the requirements. This should include start and finish dates, recruitment target, equipment and training requirements. The appendices detail any financial provisions, how materials will be shared and data processing/sharing arrangements. Items marked with a ^ are to be completed by the practice; then the OID should be returned by email from an authorised signatory (wet ink signatures are not expected). The Local OID is not a final document presented to the practice, but is to be negotiated and agreed between the practice and the study. Here is where you can agree targets, finances and local arrangements. |

An unmodified site agreement can also be used:
|
|
Unmodified agreements have been negotiated and reviewed by legal teams centrally, so practices can be assured that they can sign up to these, provided they have reviewed and agree to the study detail in the appendices. The PIC agreement is appropriate when acting as a Participant Identification Centre (PIC) [see Understanding the role of PICs guide]. It is an agreement between the practice and the research site (often a hospital trust). The person signing the agreement must be an appropriately delegated member of staff from the practice. An electronic signature will often be acceptable with agreement from the Sponsor. The signed copy should be kept in the site file (either hard copy or electronic) |
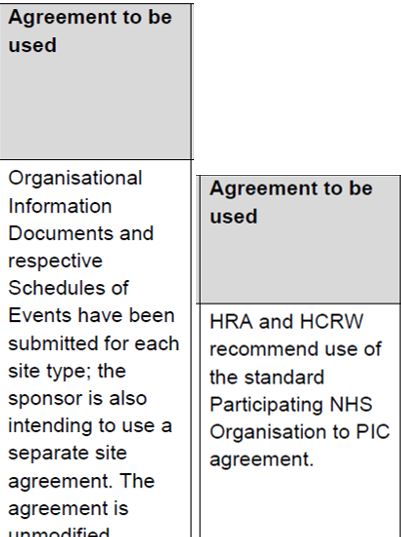
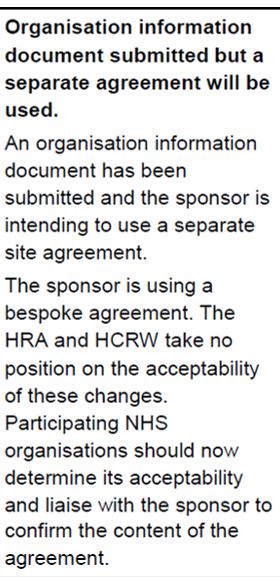
Modified/bespoke agreements are not recommended but can be used if agreed by the HRA:
|
|
It should be specified in the HRA letter where the Sponsor has used a modified or bespoke agreement. Where non-standard/modified agreements are used, the practice may wish to seek legal advice, but at the very least should carefully check through the agreement terms to make sure they are happy with these.
|
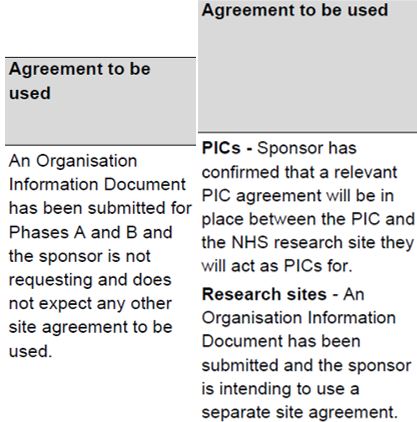
Sometimes, different parts of the study are subject to different agreements.
|
|
When different parts of the study are mentioned, the practice should confirm which parts of the study are applicable to them. The detail should be found in the OID, but if not sure you should clarify with the study team and receive confirmation in writing where possible. |
Other document types:
Studies which started a number of years ago may try to use older documents. The SSI (Site specific information) and Statement of Activities are now out of date so can no longer be used for confirming participation. The study team should be directed to https://www.myresearchproject.org.uk/help/hlpsitespecific.aspx#UK-Local-Information-Pack-Transition to replace these documents with the current ones.
Further advice and examples of setting up studies in primary care:
You can read more about study setup in primary care settings, including examples, at https://s3.eu-west-2.amazonaws.com/www.hra.nhs.uk/media/documents/HRA_Approval_in_Primary_Care_Settings__Principles_of_Study_Set-Up_Version_2.0_09Mar17.pdf (note that this document contains some old references but can be taken as the most recent guidance).
Glossary of Acronyms and Terms:
-
C&C - Capacity and Capability
-
RDN - Research Delivery Network
-
HRA - Health Research Authority
-
OID - Organisation Information Document
-
PI - Principal Investigator
-
PIC - Participant Identification Centre
-
SSI - Site Specific Information (form)