
Please contact us if you feel there is any information not included in these guides that it would be useful for us to add, or with any queries.
The below information was correct at the time of writing and is regularly reviewed to ensure all links are still active. However, guidance can change rapidly. We will keep reviewing this information to check information is still current, but if in any doubt please do check external sources. Please do contact us with any issues. Thank you. Last reviewed 10/09/2025.
Any printed version of this document should be considered uncontrolled. Version control is only applicable to the documents that are live on the NHS West Yorkshire Research and Development website © NHS West Yorkshire Integrated Care Board
How to guide: Getting your practice ready to do research
This guide describes the things you need to do to get your practice ready to start doing research i.e. the steps you should take before your first study.
Key information you should be aware of:
-
Practices should be aware of their responsibilities with regard to research. Practices continue to be responsible for their own patients’ care, and retain responsibility as data controller for the patient records held by the practice (see the data protection guide for more detail).
-
The practice is responsible for ongoing treatment of its patients, and for ensuring that patients and patient data are treated with due care. The practice retains responsibility as data controller for data in the practice system.
-
The practice is responsible for ensuring that all staff who may be involved in a study, have an understanding of what is involved.
-
Practice staff are responsible for ensuring oversight of all data use (see the data protection guide), ensuring that persons accessing the practice are suitably trained and only undertaking activities appropriate and approved as part of the study (see the staff guide).
-
The Clinical Negligence Scheme for General Practice is responsible for providing insurance for negligence in the delivery of research. The Sponsor insurance is responsible for insuring any problems with the design of the research.
-
NHS R&D departments (West Yorkshire R&D for all GP practices in West Yorkshire) and the NIHR RDN (Research Delivery Network) are responsible for advising practices with regard to their participation in research.
-
Sponsors are responsible for the initiation, management and financing of a specific study.
-
Chief Investigators are responsible for the conduct of a specific study within the UK.
-
Principal Investigators are responsible for the conduct of a specific study at a research site. Tasks may be delegated but responsibility remains with the Principal Investigator.
-
Practices are responsible for ensuring an appropriate privacy notice is displayed within the practice and is on the practice website.
Highlights
-
Consider what types of research you want to do, and what you can do
-
Get your staff ready and aware of what they need to do
-
Consider appointing a research lead
-
Prepare a research file and documents
-
Get contracts in place
-
Advice and support are available from the Research Delivery Network (RDN) and West Yorkshire Research and Development (WY R&D)
-
Infrastructure funding is available from the RDN
See also
-
Data protection guide
-
Research training guide
-
Roles and terminology guide
-
Finance guide
-
Staff guide
Full guide
Considering what you want to do:
Practices should consider what types of research projects are of interest, and how much time and resource they are able to make available for research. As a guideline, practices may consider:
-
Does the practice only want to get involved with studies adopted by the National Institute of Health Research portfolio (see definitions)? These studies may include service support costs (see definitions). Non-portfolio studies may also include payments.
-
Are there particular topic areas of interest?
-
Are there particular areas where the patient population is likely to be of interest to researchers? For example a high prevalence of a particular condition?
-
Is the practice only able to consider participating in research where their time will be funded?
-
Is the practice only interested in participating as a Participant Identification Centre (PIC) (a PIC is where the practice participates only to identify and invite participants, but does not receive consent or conduct any research activity)?
-
What staff time is the practice able to offer? Admin staff time? GP time? Nurse time? Or simply practice space for external researchers to do research?
-
What facilities do the practice have which could be of interest to a research study (e.g. medicine storage, equipment such as specialist scanners, freezers)?
-
Is a member of practice staff willing to act as a Principal Investigator (PI) - responsible for research at the site? Many studies require sites to appoint a PI.
Getting your staff ready and aware of what they need to do:
-
Practices should consider appropriate training for research, which may include RDN Fundamentals of research training, Good Clinical Practice, Informed Consent training, and study-specific training. All available for free through NIHR Learn (registration required).
-
Practices are advised to communicate with all practice staff who may be involved, to ensure everyone is aware of the reasons for doing research, what may be required of them, and things they should look out for. Alternatively the RDN or West Yorkshire Research and Development may be able to present to staff.
Staffing:
-
Practices should identify a lead for research, who takes responsibility for overseeing the study and liaising with the study team.
-
Practices should consider appointing a research nurse or other specialist.
-
Practices should ask all involved staff (at any level of involvement) to complete a research CV. The template research CV can be found here.
-
Practices should consider identifying deputies for key research roles to ensure cover in case of illness or annual leave.
Preparing a research file and documents:
-
Research file/folders: We would recommend that your practice begin a research file (electronic and/or paper). This could include GCP certificates and any other research related information, for example any interest you have expressed in specific studies, which will be useful if you apply for start-up or cluster funding at some point. Here is a suggested electronic file arrangement you may wish to create:

-
You will need a secure place where confidential study documents (e.g. signed consent forms) can be stored.
-
Both the electronic and paper file storage should be accessible to staff working on the study but able to be kept confidential to anyone else.
Getting contracts in place:
-
Practices will need to hold a contract with the Regional Research Delivery Network in order to receive any service support cost payments for research studies. This should be requested by your local contact in the RDN.
-
Practices may consider applying for Research Ready status from the Royal College of GPs.
Advice and support are available from the Regional Research Delivery Network (RDN) and West Yorkshire Research and Development (WYRD):
-
Practices are advised to liaise with the Research Delivery Network and West Yorkshire Research and Development, or equivalents in other areas, for advice about starting to do research. Advice can also be found on the West Yorkshire Research and Development website, the HRA website, the NIHR RDN website and the RCGP website.
-
Practices may consider applying for infrastructure funding from the RDN (when available).
Processes:
-
Practices should agree a process for researchers and RDN nurses who may access the site – ensuring that they know appropriate fire and health and safety procedures and receive badges/access fobs/smartcards as required (see the staff guide).
Informing patients:
-
You must make sure patients are aware that patient records may be accessed for use in research, and shared with other organisations for that purpose. This means displaying a privacy notice on the practice website, physically in the practice, and within any practice materials given to patients when they join the practice. The statement should mention that patients can opt out. You should share this information as widely as possible, to ensure patients have the opportunity to see the information and object if they wish. There is advice available from the BMA on privacy notices. See also the data protection guide.
-
Your Patient Participation Group (PPG) should be aware and supportive of your drive to get ready to do research. They can also help to suggest what research might be welcomed by your patients.
-
Where practical, we would recommend making available a notice board or an area for research within your waiting room(s). If you have access to add material to the information displayed on the screens in your waiting rooms, you could consider including some slides about research. If you would like us to supply slides, please contact West Yorkshire R&D.
Other initiatives to consider:
-
You can promote Join Dementia Research to your patients – all the practice needs to do is promote the initiative and patients engage directly with researchers.
-
Clinical Practice Research Database is a system which extracts anonymised data from your patient record system for use in approved research. Practices can learn more and sign up here: link
-
The RCGP surveillance centre is a similar system which extracts pseudonymised data from the patient record system and also provides a dashboard back to the practice to help with Quality Improvement. Practices can sign up here: link
Glossary of Acronyms and Terms
-
BMA - British Medical Association
-
GCP - Good Clinical Practice training
-
PI - Principal Investigator
-
PPG - Patient Participation Group
-
RCGP - Royal College of General Practitioners
-
RDN - Research Delivery Network
-
SIV - Site Initiation Visit
-
SOPs - Standard Operating Procedures
How to guide: Training your practice staff to support research delivery
This guide outlines the training available to prepare your practice staff to support the delivery of research at your practice.
Key information you should be aware of:
Practice staff should be aware of their responsibilities with regard to research and be adequately trained to carry out the task(s) delegated to them. Practices continue to be responsible for their own patients’ care. Please note that some very simple studies (such as ones which require the practice to send a text message to patients inviting them to participate in a survey) may not require staff to complete all the training mentioned below.
-
The practice is responsible for ongoing treatment of its patients, and for ensuring that patients and patient data are treated with due care.
-
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what is involved.
-
Some studies will require that a Principal Investigator (PI) be appointed at the practice. PIs are responsible for the conduct of a specific study at each research site (the GP practice). Tasks may be delegated but responsibility remains with the PI.
-
All staff involved in research at the practice must first have their specific task(s) approved by the PI, indicated by an entry on the Delegation Log.
-
Clinical trials of investigational medicinal products (CTIMP) require that staff involved have completed NIHR Good Clinical Practice (GCP) training. While this is not a requirement for non-CTIMP studies, some studies may still request this. Check the protocol and/or with the study team for clarification.
-
All staff involved in research at the practice must be familiar with latest protocol requirements and Standard Operating Procedures (SOPs).
-
All staff involved in research at the practice must have completed study specific training where this is required for the study.
-
NHS R&D departments and the Regional Research Delivery Network (RRDN) are responsible for advising practices about their participation in research.
-
The Clinical Negligence Scheme for General Practice is responsible for providing insurance for negligence in the delivery of research. The Sponsor insurance is responsible for insuring any problems with the design of the research.
-
Sponsors are responsible for the initiation, management and financing of a specific study.
-
Chief Investigators (usually appointed by the sponsor) are responsible for the conduct of a specific study within the UK.
Highlights
-
General training for practices
-
Training for staff involved in consenting participants
-
Study specific training
-
Protocol and SOPs
-
Delegation of duties
-
Keeping up-to-date
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Data protection guide
Full guide
General training for practices:
-
An Introduction to Research Delivery in Primary Care (offered by West Yorkshire Research and Development):
-
This free 20-minute training session provides a basic overview of research delivery, addresses some common concerns and includes a Q&A section.
-
It is intended for all practice staff supporting research in some way, if they will not be receiving consent from patients. This would generally include practice managers, admin staff, receptionists, practice nurses and others not involved in the consent process.
-
To arrange this for your practice, please email wyicb-bdc.research@nhs.net.
-
A video entitled: GP Practices: Shaping the Future of Research has been produced by West Yorkshire R&D and the NIHR Yorkshire and Humber Regional Research Delivery Network. This 13-minute video is similar in content to the above Introduction to Research Delivery in Primary Care training session, but it can be viewed at any convenient time for practice staff. The video may be viewed via this link: GP Practices Shaping the Future of Research - YouTube
-
Good Clinical Practice (GCP) training
-
All CTIMP studies and some non-CTIMP studies (where stipulated in the study protocol or requested by the study team) will require that staff complete GCP training:
-
Enrol for GCP training via the National Institute for Health Research (NIHR) Learn website https://learn.nihr.ac.uk.
-
A certificate should be downloaded and saved by the member of staff completing this training.
-
Staff members who will be carrying out any tasks requiring a GCP certificate for a study should ensure a copy of their GCP certificate is in the Site File for the relevant research project (see the site file guide for more information).
-
While there is no specific expiry for GCP training, some studies do have a requirement that it be up-to-date. Therefore, it is generally recommended that GCP refresher training is completed every 2-3 years via the National Institute for Health Research (NIHR) Learn website: https://learn.nihr.ac.uk.
-
Study specific training:
-
For many studies (with the exception of very simple studies) the study team will provide study specific training. This is sometimes part of a Site Initiation Visit (SIV). SIVs may be in person or virtual:
-
It is important to ensure all staff who will be involved in the study attend study training including the SIV where applicable. If this is not possible, pertinent information should be shared with involved staff as soon as possible.
-
Staff should feel free to ask questions of the study team if anything requires clarification.
-
If there is a Training Log in the Site File (see Site File guide for more information), an entry should be made for each staff member who attends any study specific training such as SIVs.
-
Protocol and Standard Operating Procedures (SOPs):
-
The latest version of the Protocol and any SOPs should be kept in the Site File (see Site File guide for more information):
-
It is the responsibility of the PI to ensure the latest version of these documents is in the Site File.
-
All staff with any assigned tasks associated with the research project must be familiar with the requirements of their task(s) as outlined in the latest version of the Protocol and SOPs.
-
Delegation of duties:
-
For many studies (with the exception of very simple studies) the Site File should contain a Delegation of Duties Log (Delegation Log). A delegation log delegates specific duties to specific members of the team.
-
The delegation log template should be provided by the study team, although it’s the responsibility of the PI to complete and maintain the Delegation Log.
-
Where a Delegation Log exists:
-
All staff involved in the research project at the site (GP practice) should only carry out tasks that they have been delegated to carry out by the PI. The staff member should be named on the Delegation Log with their particular task(s) noted.
-
Before agreeing to any task, all staff members should ensure that they are appropriately trained to carry out that task.
-
Staff listed on the Delegation Log should not carry out any task(s) until this has been approved and signed off by the PI.
-
Activities should have a start date and, when the person stops undertaking the duty, it should have an end date.
-
Keeping up-to-date:
-
During the course of a research project, the study team may make modifications/amendments to study processes and/or documentation. Practices must comply with any modifications and ensure they are working to the latest procedures and documents. Modifications may include changes to the following:
-
Documents such as the Protocol and SOPs, and participant facing documents:
-
See Site File guide.
-
All staff should be made aware of the changes and ensure that they complete any assigned tasks in line with adjustments made in these documents.
-
-
Staff:
-
Where a new member of staff will be carrying out tasks related to a research project, they should be appropriately trained in line with this guide.
-
All staff should be made aware of the changes and ensure that they complete any assigned tasks in line with adjustments made in these documents.
-
-
Glossary of Acronyms and Terms:
-
CI Chief Investigator
-
CTIMP Clinical trials of investigational medicinal products
-
GCP Good Clinical Practice training
-
PI Principal Investigator
-
RRDN Regional Research Delivery Network
-
SIV Site Initiation Visit
-
SOPs Standard Operating Procedures
How to guide: Understanding the role of PICs
This guide outlines key information about the role of the Participant Identification Centre (PIC). PICs are NHS/HSC organisations that identify potential research participants to be recruited at a separate research site, should they wish to participate.
Key information you should be aware of:
Practice staff should be aware of their responsibilities with regard to research and should only take part in approved studies. Practices continue to be responsible for their own patients’ care but PICs are not research sites and are not treated in the same way as research sites. Practices should understand study requirements and what can and can’t be done by reviewing the study protocol and Organisational Information Document (OID) and should ensure that data protection guidance is followed.
-
PICs are not considered to be research sites but should put in place an appropriate agreement to participate
-
PICs retain responsibility for the healthcare of patients outside of research but research sites have duty of care for participants in relation to research studies
-
PICs should assess studies in relation to practice capacity and capability, and the appropriateness of participant identification activities
-
Data protection needs to be given consideration
Highlights
-
What is a PIC?
-
Overview of the role of PICs
-
Things to consider
-
Contracting arrangements
Full guide
What is a PIC?:
Participant Identification Centres (PICs) are NHS/HSC organisations that identify potential research participants.
A GP practice is operating as a PIC if its role involves:
-
identifying potential research participants by processing personal data (e.g. searching patient record databases);
-
it is following sponsor instructions in identifying potential research participants;
-
the practice then directs potential participants to a separate research site without undertaking further research activity for a study.
Overview of the role of PICs:
Some studies recruit participants via general practices but then any further research activity takes place in another setting e.g. a hospital. These are known as Participant Identification Centre (PIC) studies.
If a GP practice agrees to be a study PIC then the study team does not have permission to carry out research activity at the practice but general practices can be involved in supporting recruitment to studies by identifying participants. This can involve:
-
conducting a search of a database to identify a list of participants who fit a study’s inclusion/exclusion criteria;
-
sending out an invitation to patients to introduce a study e.g. a letter, email or text.
A practice is not acting as a PIC if:
-
informed consent of the participant is taken by practice staff;
-
any assessment outlined in the study protocol to determine whether potential participants are eligible for the research study (e.g. a screening blood test or x-ray) is carried out by the practice, or any aspect of research delivery outlined in the protocol is carried out by the practice;
-
potential research participants are referred to a study as part of normal clinical activity (e.g. routine clinics) in order to gain access to clinical interventions.
Advertising opportunities to participate in studies e.g. via a poster in a waiting room, is not PIC activity, so any practice can do this without a formal agreement.
If the study requires the health care professional to consent patients or carry out any other research activity (e.g. follow-up tests) then it is should be treated as a full research study and not as PIC activity.
PIC activity for NHS organisations in England may only commence once:
-
a study has received HRA Approval;
-
the research site linked to the PIC has completed its capacity and capability assessment or other review as appropriate;
-
there is an appropriate agreement in place between the research site and the NHS organisation acting as a PIC.
Things to consider
If you are considering whether to be a study PIC you should consider:
-
Does the patient information clearly detail who is delivering the research?
-
Is HRA approval in place and has West Yorkshire R&D (or your local R&D team) had sight of this study?
-
Have you considered the finance and resource implications of being a PIC? Some service support costs (SSC) and research costs may be reimbursed by the NIHR Clinical Research Network or the study team to cover, for example, postage costs or the time taken by practice staff to carry out a search.
Contracting arrangements
As per IRAS guidance: NHS/HSC PICs should be set up by through a sub-contracting arrangement with the participating NHS/HSC organisation that the PIC supports. Appropriate data processing arrangements should be put in place by using the appropriate agreement. Best practice is for studies to use a model agreement:
-
model Non-Commercial Participant Identification Centre Agreement (mNC-PICA) - Trial Site to PIC
-
model Non-Commercial Participant Identification Centre Agreement (mNC-PICA) - Sponsor to PIC
Source: https://www.myresearchproject.org.uk/help/hlpsitespecific.aspx
Glossary of Acronyms and Terms
-
NHS - National Health Service
-
HSC - Health and Social Care Services
-
PIC - Participant Identification Centre
-
HRA - Health Research Authority
-
OID - Organisational Information Document
How to guide: Understanding roles and terminology
This guide outlines the roles and terminology commonly used in research.
Key information you should be aware of:
Practice staff should be aware of their responsibilities with regard to research, and should understand both research roles and commonly used research terminology.
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what is involved:
-
Staff involved in the consenting of patients may need to complete Good Clinical Practice (GCP) training. This is a legal requirement for Clinical Trials of Investigational Medicinal Products (CTIMPs) and may be a requirement specified by the study protocol.
-
All staff involved in research at the practice must have completed study specific training where this is required for the study.
-
Check with the study protocol what is required of staff.
Highlights
-
Roles
-
Individual roles
-
Broader roles
-
Terminology
-
Research sites
-
Research study documents
-
Other
See also
-
Getting your practice ready to do research guide
-
Research training guide
Full guide
Roles:
Chief Investigator (CI)
The chief investigator is the overall lead researcher for a research project. In addition to their responsibilities if they are members of a research team, chief investigators are responsible for the overall conduct of a research project:
-
The Chief Investigator’s responsibilities are set out in more detail in the UK Policy Framework for Health and Social Care Research.
Principal Investigator (PI)
An individual responsible for the conduct of the research at a research site. There should be one PI for each research site. In the case of a single-site study, the chief investigator and the PI will normally be the same person. The PI may delegate duties to other staff, but will retain responsibility
Research Sponsor
The organisation or partnership that takes on overall responsibility for proportionate, effective arrangements being in place to set up, run and report a research project:
-
All health and social care research should have a sponsor. This includes all research that involves NHS patients, their tissue or information.
-
Two or more organisations may agree to act as co-sponsors or joint sponsors. Co-sponsors allocate specific sponsor responsibilities between them whilst joint sponsors each accept liability for all of the sponsor’s responsibilities.
-
A sponsor can delegate specific tasks to any other individual or organisation that is willing and able to accept them.
-
Any co-sponsorship, joint sponsorship or delegation of tasks to another party should be formally agreed and documented by the sponsor(s).
-
The sponsor’s responsibilities are set out in more detail in the UK Policy Framework for Health and Social Care Research.
Collaborator
An organisation other than the sponsor that provides support for a clinical study. This support may include activities related to funding, design, implementation, data analysis, or reporting.
Local collaborator
A person undertaking certain types of straightforward research procedure, not requiring the appointment of a Principal Investigator and/or a site agreement. A named local collaborator may be required at a site, which will be specified in the HRA approval letter.
Study management and monitoring
Various groups may be set up to manage, oversee and guide a study throughout its lifetime. These may include representatives from the sponsor institution, from those delivering the study, from patient groups and from other guiding or advisory members. These may include:
Trial Steering Committees or Study Steering Committees
All primary research projects are required to establish a Trial Steering Committee (TSC) or Study Steering Committee (SSC). The role of the TSC/SSC is to provide overall supervision for a project on behalf of the Research Sponsor and Research Funder and to ensure that the project is conducted to the rigorous standards set out in the Department of Health’s UK Policy Framework for Health and Social Care Research and the Guidelines for Good Clinical Practice. It should be noted that the day-to-day management of the project is the responsibility of the Chief Investigator, and as such the Chief Investigator may wish to set up a separate Project Management Group (PMG) to assist with this function.
Data Monitoring Committee (DMC)
A group of independent scientists who monitor the safety and scientific integrity of a clinical trial. The DMC can recommend to the sponsor that the trial be stopped if it is not effective, is harming participants, or is unlikely to serve its scientific purpose. Members are chosen based on the scientific skills and knowledge needed to monitor the particular trial. Also called a data safety and monitoring board, or DSMB.
Terminology – Research sites:
It is important to note that you may be asked to put up a poster to promote a research study (or advertise e.g. via social media). You can do this without being any of the site types below:
Research Site
A research site is responsible for research activities, such as:
-
Any protocol-specified assessment to determine whether the potential participant is eligible for the research study (e.g. a screening blood test or x-ray).
-
The recruitment (informed consent) of participants into the research study.
-
The delivery of research activities and procedures as specified in the research protocol.
-
Providing data from the clinical records.
Participant Identification Centre (PIC) site
PICs are National Health Service (NHS) or Health and Social Care (HSC) organisations that identify potential research participants. They are not research sites. An NHS/HSC organisation is operating as a PIC when it meets the following three criteria:
-
Identifies potential research participants by processing personal data (e.g. through carrying out a search of a patient records database to identify individuals that meet a study’s eligibility criteria).
-
Is following the sponsor(s)/protocol instructions in identifying potential research participants.
-
Directs these potential participants elsewhere without undertaking any further research activity for that study (i.e. the research occurs at another organisation).
See also the section for understanding the role of PIC sites
Multicentre trial/study
A trial or study conducted at several geographical sites; trials are sometimes conducted among several collaborating institutions, rather than at a single institution - particularly when large numbers of participants are needed.
Lead site
In the case of a multi-site trial/study, the site for which the Chief Investigator is also the Principal Investigator.
Terminology - Research documentation:
It is important to note that the study documents listed below will have been scrutinised and approved by an Ethics committee during the Health Research Authority (HRA) approval process. It is important that only approved versions of documents are used and no changes are made without an amendment to the HRA:
Protocol
A document that describes the objectives, design, methodology, statistical considerations (or other methods of data analysis) and organisation of a research study.
Patient information sheet (PIS) or Patient information leaflet (PIL)
Researchers must provide a patient information leaflet to everyone they invite to take part in a research study, to ensure people can make an informed decision about this. The PIS explains what taking part will involve and should include details about: why the research is being done, how long it will last, and what methods will be used; the possible consequences; contact details; how the results will be shared with others.
Consent form
A consent form (sometimes called ICF or informed consent form) must be used to record the consent process and a participant's agreement to take part in a research study. When producing the consent form consideration should be given to what is appropriate for the type of study and the participants who will be involved.
Delegation log
The delegation log provides clarity regarding who is responsible for undertaking what activity during delivery of the study. The delegation log is a tool to be maintained throughout the life time of the study at that participating research site. Research activities are delegated by the Principal Investigator (PI).
Case report form (CRF)
A case report form (CRF) is designed to collect the patient data in a clinical trial. It can be a printed paper or an electronic document. It is designed to record all of the protocol required information to be reported to the sponsor for each trial participant. The size of a CRF can range from a one-time 'snapshot' of a patient's physical condition to hundreds of pages of electronically captured data obtained over a period of weeks or months. It can also include required follow-up visits months after any intervention or treatment has stopped.
Organisational information document (OID) and schedule of events costing attribution template (SoECAT)
If a study is non-commercial, the sponsor of the study is responsible for preparing an organisational information document (OID) and a schedule of events cost attribution template (SoECAT). These documents capture all the information around study activities to be undertaken at a local level. A template OID and SoECAT will be reviewed and approved by the HRA for each type of participating site to ensure that there is clarity on the resource implications for participating NHS organisations delivering the research.
Model agreement/contract
The HRA provides model agreements for many types of trial, and the agreement being used for sites will be approved as part of the HRA approval and listed in the HRA approval letter. The HRA prefers that unmodified model agreements are used, with only the addition of the study specific information. You can be assured that unmodified model agreements have been checked and approved as part of the HRA approval process.
If the HRA agrees that another form of agreement may be used, this will also be specified in the HRA approval letter. However the HRA usually states that it doesn’t vouch for such agreements, and the practice may wish to seek legal advice before signing.
Terminology – Other:
If you would like to understand more terminology used in research the National Institute for Health Research (NIHR) provide a useful glossary: https://www.nihr.ac.uk/glossary
How to guide: Responding to Expressions of Interest (EoIs)
This guide outlines the process for expressing interest in specific studies and outlines some important points to consider before expressing interest. The term Expression of Interest (EoI) can refer to the information a practice receives about a study, as well as interest in a study as expressed by the practice. EoIs (usually in the form of an email) may be sent to a practice by West Yorkshire Research and Development, the Regional Research Delivery Network (RRDN), or direct from a study team.
Key information you should be aware of:
Please note that expressing interest in a study does not commit the practice to participate; nor does it guarantee involvement. If you receive information about a study direct from a study team, and the study is recruiting via the NHS, you will need to ensure all appropriate approvals are in place. This would include Health Research Authority (HRA) approval and, in most cases, approval by a Research Ethics Committee (REC). Both of these approvals are in the form of a letter which should be available from the study team. Where the information about a study comes from West Yorkshire Research and Development or the RRDN, these approvals should already be in place.
-
The practice is responsible for ongoing treatment for its patients, and for ensuring that patients and patient data are treated with due care.
-
Most studies will require that a Principal Investigators (PI) be appointed at the practice. PIs are responsible for the conduct of a specific study at each research site (the GP practice). Tasks may be delegated but responsibility remains with the PI (see Appointing a Principal Investigator guide for more information).
-
Many studies require that staff who will be involved in recruiting participants and receiving consent must have completed NIHR Good Clinical Practice (GCP) training. Check the protocol and/or with the study team for clarification.
-
NHS R&D departments and the Research Delivery Network (RDN) are responsible for advising practices with regard to their participation in research.
-
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what is involved.
-
The Clinical Negligence Scheme for General Practice is responsible for providing insurance for negligence in the delivery of research. The Sponsor insurance is responsible for insuring any problems with the design of the research.
-
Sponsors are responsible for the initiation, management and financing of a specific study.
-
Chief Investigators are responsible for the conduct of a specific study within the UK.
Highlights
-
Assessment of Feasibility
-
Submitting EoIs
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Site Initiation Visit guide
-
Data Protection guide
Full guide
Assessment of feasibility:
Before expressing interest in a study, the practice(s) must first consider whether the study is feasible for the practice(s).
-
Note that some studies require minimal involvement from practices (such as surveys, questionnaires and interviews). If this is the case, a detailed consideration of feasibility may not be required.
There is usually a deadline for expressing interest, and at times, this is a very tight deadline. Therefore, it may be helpful to have a checklist to which you may refer to help you assess feasibility. If the information about the study has come via West Yorkshire Research and Development or the RDN, an overview of the information you need should be in the EoI email. You may need to read the Protocol, Organisation Information Document or Participant Information Sheets for more information.
Below are some suggested points you may wish to consider when determining feasibility. This is not necessarily an in-depth feasibility analysis, but more of a surface review. Please also note that this is not a comprehensive list, and there may be other points to consider based on local circumstances. Therefore, it may be good to create your own checklist, as suggested above:
STAFF: Does the practice have staff available to deliver the study if required?
-
Are they appropriately trained or experienced to do this (see the Research Training guide for more information)? Note that the study team may provide study specific training.
-
Does the study require that a Principal Investigator be appointed at the practice and if so, is someone willing, available and appropriately trained and/or experienced to do this (see also PI Guide)? The study may require this to be a GP or a nurse.
-
Is there sufficient interest and enthusiasm for research among practice staff who will be involved?
FACILITIES: Does the practice have the appropriate facilities to deliver the research? For example, this may include one or more of the following:
-
Clinic room(s).
-
Safe data storage (electronic or paper). See also the Data Guide.
-
Equipment (sometimes, some or all of this may be supplied by the study team).
-
Are there any IT requirements, such as the installation of some software? If so, will this be done by the practice, the local IT provider or the study team? Is such an installation possible in view of firewalls and security? Please feel free to check with West Yorkshire Research and Development (WY R&D) and/or your IT provider since some prior work may have already been done to allow this within the IT network. It is often the case that practices cannot install software themselves, so engaging with the IT provider and/or WY R&D will often be required.
COSTS: Service Support Cost (SSC) payments may be provided to the practice via the RDN, Research Costs may be provided by the study team. Some studies also include Excess Treatment Costs (costs for treatments over and above standard care). These are paid quarterly in arrears via the national payment system to the site where they are incurred.
TIME: Does the practice have the time to carry out the local requirements of the study? (Note: some time may be reimbursed by means of Service Support Costs).
PATIENTS: Is there a specific recruitment target (number of patients recruited to the trial) for the practice? If so, does the practice have the sufficient number of patients on their clinic list which meet the inclusion criteria for the study? It is generally expected that 10% of patients approached about a study may wish to participate.
-
Is there another study being delivered at the practice which is already recruiting patients with the same characteristics, or has one been delivered recently? If so, will it be appropriate for these patients to be approached again?
-
Is the study relevant to the patient population of the practice? While there is a need to avoid making assumptions, it may be easier to recruit patients to a study if this is relevant to them.
Submitting EoIs:
Once a practice has determined that a study is feasible to be delivered at the practice(s), an expression of interest (EoI) should be made promptly. There is often a short deadline for this.
-
Please be sure to send your EoI to the contact supplied in the EoI email.
-
Save a copy of your EoI. Not all practices may be selected for involvement in a study, since the study team may only require a specific number of practices, or practices with specific criteria and/or previous research experience. Therefore, it may be useful to have a list of EoIs sent by the practice as evidence for any applications for research funding which may be made by the practice.
-
Wait for a response from the study team, the RDN or West Yorkshire Research and Development before implementing anything related to the study.
-
Please note that for some studies, a response may not be received. This should be understood to mean that the practice has not been selected to deliver the study.
Glossary of Acronyms and Terms:
-
CRN - Clinical Research Network
-
GCP - Good Clinical Practice training
-
PI - Principal Investigator
-
SIV - Site Initiation Visit
-
SOPs - Standard Operating Procedures
How to guide: Site selection / initiation visit (SIV)
Key information you should be aware of:
Please note that expressing interest in a study does not commit the practice (site) to participate; nor does it guarantee involvement.
-
Some studies may not require the study team to confirm that the practice can be involved (such as a study which simply asks a practice to send patients and/or staff a link to a survey).
-
Other studies may require that individual practices be selected to participate by the study team.
-
Where a study team selects a practice to participate in a study, a Site Initiation Visit (SIV) is usually arranged.
-
The practice is responsible for the ongoing treatment of its patients, and for ensuring that patients and patient data are treated with due care.
-
Most studies will require that a Principal Investigators (PI) be appointed at the practice. PIs are responsible for the conduct of a specific study at each research site. Tasks may be delegated but responsibility remains with the PI.
-
Many studies require that staff who will be involved in recruiting participants and receiving consent must have completed NIHR Good Clinical Practice (GCP) training. Check the protocol and/or with the study team for clarification.
-
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what this involves.
-
Sponsors are responsible for the initiation, management and financing of a specific study.
-
Chief Investigators (CIs) are responsible for the conduct of a specific study within the UK.
See also
-
Getting your practice ready to do research guide
-
Research training guide
-
Roles and terminology guide
-
Data protection guide
Full guide
Site selection:
Some studies may not require the study team to confirm that the practice can be involved (such as a study which simply asks a practice to send patients and/or staff a link to a survey). This will usually be made clear in the Expression of Interest (EoI) email you receive from the Research Delivery Network (RDN), from West Yorkshire Research & Development (WY R&D) or from the study team. If in doubt, please feel free to ask the question of the sender of the EoI.
Other studies may require that individual practices be selected to participate by the study team. Depending on the complexity of a study, this may be based on one or more of the following:
-
Demographics of the practice’s patient population (i.e. sufficient numbers of potential participants meeting the inclusion criteria for the study)
-
Previous research experience of the practice
-
Previous recruitment figures demonstrated by the practice
-
Staff skills, experience and training
-
Availability of a PI
-
Other factors specific to the study
If site selection is required for a study, do not start any study procedures until this has been confirmed by the study team.
Due to the number of EoIs received by study teams, they may only reply to practices that are selected to participate. Therefore, no reply can usually be assumed to mean that your practice has not been selected for a particular study on that occasion.
Even if your practice is not selected to participate, it would be good to save a copy of the interest expressed. This could be helpful to establish intent and interest, for example when applying for future research infrastructure funding.
Site Initiation Visit (SIV):
-
Where a study team selects a practice to participate in a study, a SIV is usually arranged. This could be in the form of a telephone call, video conference, or face-to-face meeting.
-
The study team should tell you who will need to be present for the SIV, but you should also make sure all relevant staff are included from the practice point of view.
-
The SIV is not only an opportunity for the study team to explain details of the study, provide necessary training and give direction as to completing the delegation log etc., but it is also an opportunity for staff members to ask questions and raise concerns.
-
Attendance at a SIV should be documented and could be used in staff appraisals.
How to guide: Delegation of duties log (or delegation log)
Key information you should be aware of:
The delegation log (stored in the Site File, usually alongside research CVs and GCP certificates) is a list of study-related duties assigned to staff members It is a live document which must be kept up to date if staff working on the study change roles or leave etc. The Principal Investigator is responsible for duties carried out in connection with the study, but the delegation log enables them to delegate some specified duties to other staff. Staff members must only carry out duties to which they are assigned on the delegation log. Please note that some very simple studies (e.g. surveys) may not have a delegation log. If in doubt, staff should always check with the study team before handling any study-related task.
-
The practice is responsible for ongoing treatment for its patients, and for ensuring that patients and patient data are treated with due care.
-
Most studies will require that a PI be appointed at the practice. PIs are responsible for the conduct of a specific study at each research site (e.g. the GP practice). Tasks may be delegated but responsibility remains with the PI.
-
Many studies require that staff who will be involved in recruiting participants and receiving consent must have completed NIHR Good Clinical Practice (GCP) training. Check the protocol and/or with the study team for clarification.
-
NHS R&D departments and the RDN are responsible for advising practices with regard to their participation in research.
-
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what this involves. The study team will ensure any study specific training is provided (this should be detailed in the OID).
-
The Clinical Negligence Scheme for General Practice is responsible for providing insurance for negligence in the delivery of research. The Sponsor insurance is responsible for insuring any problems with the design of the research.
-
Sponsors are responsible for the initiation, management and financing of a specific study.
-
Chief Investigators are responsible for the conduct of a specific study within the UK.
Full guide
Delegation of duties log (or delegation log)
-
A delegation log should exist for studies, apart from very simple studies (e.g. surveys).
-
Blank delegation logs are usually supplied by the study team as part of the Investigator Site File (ISF). As an example, a template to a delegation log is available here: https://myresearchproject.org.uk/help/help%20documents/Signature_And_Delegation_Log_Template_v1-2.docx
-
The name of each staff member who will carry out a study-related task must appear on the delegation log. Alongside their name, the reference for the specific task(s) should be clearly marked. This should be initialled by the staff member and signed and dated by the PI.
-
The PI is responsible for ensuring that any staff member listed on the delegation log is sufficiently trained, qualified and/or experienced to carry out the task to which they are assigned.
-
The delegation log should be kept up to date, with start and end dates for staff that have been assigned and/or relieved of specific tasks. An end date should be entered if a staff member leaves the practice.
-
Some study teams may ask for up to date copies of the delegation log. This should be sent promptly when requested.
-
Additional pages of a delegation log should be clearly marked as such so that it is clear how many pages there are (e.g. 1 of 3).
-
As with all research documentation, any corrections on the delegation log should be marked with one straight line through the error. The person making the correction should initial and date the correction. Nothing should be deleted by being marked illegible or with correction fluid.
How to guide: Site file maintenance
Most studies will require that a site file be held and maintained by each recruiting site. Site files are often provided by the study team and may be in paper or electronic format.
Key information you should be aware of:
A site file must contain current versions of all documents being used in the study. Previous versions must be kept in the file but crossed through and marked ‘superseded’.
-
The site file must also contain delegation logs and research CVs for all persons listed in the delegation log.
-
Site files may be checked by sponsors or study monitors at any time.
-
Site files may be held electronically if agreed by the study sponsor.
-
Site files must be kept securely, but accessible by persons delivering the study for reference.
Highlights
The National Institute for Health Research (NIHR) provides a comprehensive list of suggested site file contents: https://www.nihr.ac.uk/documents/suggested-investigator-site-file-contents/11537
Full guide
Contents
Site files are often provided by the study team and may be in paper or electronic format.
The NIHR has provided a comprehensive list of suggested investigator site file contents, recommending the inclusion of a contents page, and advising that each section should be signed and dated upon completion: https://www.nihr.ac.uk/documents/suggested-investigator-site-file-contents/11537:
Please note, this is a suggested contents list. Please discuss a study’s site file with the study team; they will often provide the site file and request that the site keep this maintained. The NIHR website gives the following suggested list of what you might want to keep in a site file (if not provided by the study team):
-
Section 1 - Protocol / amendments - to include: current protocol, protocol amendments, historical protocols
-
Section 2 - Sample CRF/ QLQ Diary Cards. Note: If too bulky to put in file place file note in this section stating where it can be found.
-
Section 3 - Regulatory approval documentation
-
Section 4 - Site signature /responsibility log
-
Section 5 - Curriculum Vitae. Note: CVs for all research personnel listed in the signature/responsibility log should be included.
-
Section 6 - Patient Identification form and Patient recruitment /screening form
-
Section 7 - Sample of current and all historical Patient Information / Informed Consent form and GP Letter. Completed patient Information and Informed Consent Forms.
-
Section 8 – Correspondence. Note: File in chronological order all correspondence to/from the coordinating research body. File email communication. Include a separate section here for newsletters.
-
Section 9 - Minutes from the initiation meeting, monitoring logs and notes of telephone calls. Note: If the study is not monitored then state this in a file note in this section. Document telephone call in relation to agreements or significant discussions regarding trial administration, trial conduct, adverse events or protocol violations.
-
Section 10 - Blank serious adverse event forms and guidelines for their completion.
-
Section 11 - Notification of serious adverse events and/or safety reports:
o by Investigator to co-ordinating research body
o by co-ordinating research body to Investigator
o by co-ordinating research body to regulatory authorities (if this will not be supplied place a file note stating this).
-
Section 12 - Randomisation details. Note: to include instructions (if applicable).
-
Section 13 - Instructions for handling trial medication and trial related materials and shipping records. Note: This responsibility is normally that of the clinical trial pharmacist if this is the case place a file note in this section stating this.
-
Section 14 - Clinical Laboratory:
o Laboratory normal reference ranges (including revisions)
o Laboratory certificate(s)
-
Section 15 - Contracts:
o Investigator Commitment Statement/Study Acknowledgement
o Indemnity
o Confidentiality
o Clinical Trial Agreement including financial details.
o Completed and signed FDA 1572 form (if applicable)
o Financial disclosure letter (if applicable)
-
Section 16 - Investigator’s brochure and safety alert letters/Updates
-
Section 17 - Completed data queries
-
Section 18 - Study training materials
-
Section 19 - Miscellaneous (specify)
After the completion of the trial, the following must also be filed in the site file:
-
Section 20 - Investigational product(s) accountability at site. Note: This will be with the clinical trials pharmacist.
-
Section 21 - Documentation of Investigational product destruction. Note: if destroyed at site this will be with the clinical trials pharmacist.
-
Section 22 - Final report from Investigator to REC.
Section 23 - Clinical study report to document results and interpretation of trial.
Practices should liaise with study teams about the site trial requirements for specific studies.
Where a document is amended with a new version, outdated versions should be marked ‘superseded’ and placed behind the current version in the site file.
It is the responsibility of the PI to ensure the site file is maintained. The physical maintenance of the site file may be done by a staff member delegated by the PI.
Please continue to refer to the NIHR website for current guidance related to site files, and liaise with study teams if you have study specific queries.
How to guide: Appointing a Principal Investigator (PI)
Key information you should be aware of:
Most studies (except some very simple studies such as questionnaires) will require that a PI be appointed at the practice. PIs are responsible for the conduct of a specific study at each research site (e.g. the GP practice). Tasks may be delegated but responsibility remains with the PI.
Full guide
Appointing a Principal Investigator (PI)
The study team will be able to advise whether a PI needs to be appointed for the study being delivered at a practice (a PI will generally need to be appointed for all but the simplest of studies). IRAS guidance notes: ‘Principal Investigators are expected to be in place at participating NHS / HSC organisations where locally employed staff take responsibility for research procedures’.
If a PI is required for the study, there will be a need to ensure the potential PI has the appropriate skills/training to carry out the role, as well as the time and subject interest.
-
Most studies (except for very simple ones not involving consenting of participants) require the PI to hold an up to date GCP certificate. While there is no official expiry for GCP certificates, most studies require these to be within the past 2 years.
-
The study team may also provide some study specific training which they require the PI to attend.
-
The study team may require a copy of a simple research CV for the PI. This should be signed and dated. A template for this CV can be downloaded here.
-
While not a requirement, there may be some additional PI training available. Please check with WY R&D to see what might be available locally.
A PI is responsible for oversight of the study at the local site (in this case, the GP practice). Therefore, it is important that they have an understanding of what will be required and have the capacity to handle these responsibilities. This may include the following (this is not an exhaustive list, and depending on the type of study, the responsibilities may be much less):
-
Setting up and maintaining the site file
-
Carrying out the study in line with the protocol and any SOPs supplied by the study team
-
Delegating responsibilities at the practice relating to the study (completion of the delegation log)
-
Ensuring all practice staff involved in the study are aware of the site file and that those with delegated responsibilities are properly trained to do so
-
Providing data to the study team where applicable
-
Reporting Adverse Events
In some cases, a local collaborator rather than a PI is required. The guidance states that “the role of the Local Collaborator is to facilitate the presence of Sponsor / CRO [contract research organisation] research staff.” [IRAS guidance]
Glossary of Acronyms and Terms
-
NIHR - National Institute for Health Research
-
RDN - Research Delivery Network
-
GCP - Good Clinical Practice training
-
ISF - Investigator Site File (or Site File)
-
OID - Organisational Information Document
-
SIV - Site Initiation Visit
-
SOPs - Standard Operating Procedures
-
AEs - Adverse Events
-
PI - Principal Investigator
-
CI - Chief Investigator
How to guide: Assessing, arranging and confirming capability and capacity
Capacity and capability (C&C) assessment is the method by which sites consider whether they can take on a new study. In some cases this needs to be a formal assessment and confirmation, in other cases an email may suffice, or in others the Health Research Authority (HRA) may state that the study can proceed without C&C unless the site indicates any objection. Regarding C&C for Participant Identification Centre (PIC) activity, please see the ‘Understanding the role of PICs’ guide, and the below PIC section.
This guide outlines the ways in which practices should undertake the appropriate Assess, Arrange and Confirm Capacity and Capability stage of site agreement when setting up a study.
See also HRA guidance on: Assess, arrange and confirm terminology.
Key information you should be aware of:
-
West Yorkshire Research & Development (WY R&D) issues an advisory email for studies of which we have had sight, to assist practices with completing their review. Practices can request a copy of this advisory from WY R&D or from the study team.
-
Research governance is conducted centrally by the HRA. The HRA issues HRA approval which incorporates ethical review.
-
Each participating site must assess, arrange and confirm their capability and capacity to participate, dependent on the requirements of the individual study.
-
The decision to participate, and the agreement, should be at the level of the participating organisation, i.e. usually at the level of the practice (as an independent contractor). PCNs are not able to sign a contract unless they have been set up as a company, and neither a PCN nor a federation is not able to sign a contract on behalf of its members unless there is a formal agreement that they can do this. If the research is taking place at the federation level, e.g. in the out of hours service, then this could be signed off by the federation. The ICB cannot sign off on behalf of practices.
-
It is important to note that the processes confirmed in the HRA approval letter must be followed as formal confirmation or signing of agreements is legally necessary where indicated in this letter.
Highlights
-
What can happen before approval
-
Process following approval
-
How to assess
-
How to arrange
-
How to confirm
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Principal Investigator guide
-
Data guide
Full guide
Pre-HRA approval:
-
Sponsors are encouraged to liaise with potential sites as early as possible in the process of developing their study. This may be before HRA approval for the study has been received or even applied for.
-
At the pre-approval stage, sites can carry out a feasibility review of the study documents, but should be aware that the documents cannot be considered final until HRA approval is confirmed to the study team.
-
A patient could be involved to discuss study feasibility, but no patient recruitment can usually take place until the study is approved.
After HRA approval of the study:
-
The next step in the process is that the study team will receive a HRA approval letter.
-
When the HRA approval letter is received, it will confirm expectations related to confirmation of capacity and capability (in the ‘Information to support study setup’ table at the end of the letter).
-
This will include stating whether formal confirmation of C&C (capability and capacity) is required, and whether the study intends to use the Organisation Information Document (OID) as the site agreement.
-
You should ask for a copy of the letter and check this table first to inform what you need to do next.
-
WY R&D should also have the opportunity to issue an advisory email which will give advice on next steps in assessing, arranging and confirming C&C.
-
If formal confirmation of C&C is required, see below for detail of the steps.
Assess:
Further to any initial feasibility assessment done at the Expression of Interest (EOI) stage, this is the main and formal feasibility assessment. When formally confirming your capacity & capability, and/or confirming the OID or contract, this is where you commit to the study, so this assessment should be more in-depth. Depending on the study commitment, this may be quick (for example if just a staff survey), or may require more consideration.
At this stage, you should have from the study team:
-
A copy of the study protocol and local documents (the Local Information Pack).
-
A copy of the Organisation Information Document or study contract.
An advisory email will have been sent to the study team from WY R&D, and you can request a copy of this from the study team or from WY R&D.
At the 'assess' stage you should review the study protocol and/or OID/contract and consider the following:
|
Area |
Things to consider |
Specific questions to ask |
|
Suitable Investigator & Site Personal identified The Ready for Review and HRA Approval letters will confirm if a Principal Investigator (PI), a Local Collaborator or neither is required at participating organisations. Where a PI is required, the participating organisation has identified a suitable investigator. |
‘Suitable’ defined as a named investigator taking into account his/her professional qualification, knowledge of research field, expertise in the procedures involved, previous research experience, training in research methods (including informed consent), training in Good Clinical Practice (if applicable) and ability to take clinical responsibility for local research team. |
Do you have suitable staff who can give enough time to the study; can they be released to work on the study? Are they engaged and interested in the research? Do they have the required skills? Has a suitable and willing PI been appointed (from the practice team?) |
|
Conflict of Interest |
Any personal involvement of site personnel with the sponsor or funder have been declared |
Do any of the staff have any conflicts e.g. with other roles, personal relationships? |
|
Study Follow-up |
The clinical research staff or participating organisation have the capacity to support all study follow up as required |
Are you aware of the extent of follow-up activity – will there be ongoing commitments after recruitment? (e.g. follow up appointments, data from the clinical system) |
|
GCP Training |
All staff that require GCP training have accredited/appropriate training eg NIHR or similar standard |
Have the staff had GCP or other required training? |
|
Appropriate Equipment available |
Study specific equipment is on site or being provided by sponsor |
Does the study require any specific equipment and does the practice have this available? |
|
Safety |
The requirements for reporting to the sponsor on progress and pharmacovigilance |
Do you understand what reporting is required? |
|
Local arrangements for handling of Investigational Medicinal Product (IMP) |
Are there specific considerations for handling the Investigational Medicinal Product? |
|
|
Local Informed consent needs |
Obtaining consent in accordance with the study protocol while considering the needs of participants who may not adequately understand verbal explanations or information written in English |
Are there any concerns about your population engaging with the informed consent process, e.g. language needs. Any translated versions required are the responsibility of the study team. |
|
Training |
The participating organisation can accommodate training format required by the Sponsor e.g. online, face to face or routine briefings within the specified time frame required by the Sponsor |
Do you know what site initiation/training will be provided by the study team and are you able to accommodate this? (e.g. consider timing, face to face vs virtual). |
|
HR |
Local Human resource requirements to ensure that staff only undertake study specific activities that are appropriate to the job and competencies of the individual, and that appropriate supervision will be provided as required. This may include arrangements to issue letters of access or an Honorary contract where participants will be seen by non-NHS Organisation employees |
Have you reviewed the study and ensured that the staff working on the study meet the study requirements (e.g. qualified doctor, nurse, prescriber)? Are there any external staff involved in the study at the practice – these should be referred to West Yorkshire R&D for a letter of access. |
|
Recruitment Target |
Based on the projected study timelines there are sufficient numbers of potential participants meeting the inclusion/exclusion criteria when taking into account any competing trials currently open or planned to open at the participating organisation to minimise the potential for inadequate patient recruitment or non-completion of research |
How many participants does the study wish to recruit from the practice? Do you have enough eligible patients (bearing in mind that you will not manage to get every eligible patient to agree to take part)? |
|
Suitability of site |
Suitability of participating organisation facilities including; adequacy of facilities for any novel procedures or for procedures not part of existing clinical activity; availability and access to resources, facilities, equipment and storage considering impact on current levels of use for non-research activities; and considering Sponsor quality expectations. Availability of work space for sponsor team monitoring if required |
If being asked to undertake activities which are not part of standard clinical practice, are your rooms/facilities/equipment suitable? Do you have enough space/equipment so that taking part in the research will not impact on delivery of standard care? |
|
Indemnity |
The HRA ensures that the sponsor has appropriate insurance for the design and management of the research. It is then a sponsor responsibility to assure itself that each individual site has appropriate insurance for the delivery of the research. For NHS sites, this is inherent and derived from NHS Litigation Authority. For non NHS sites and independent contractors, acting as a site, they will need to arrange suitable indemnity themselves, which should be reviewed and deemed appropriate by the sponsor. |
General practices’ delivery of research is now covered by CNSGP (https://www.hra.nhs.uk/planning-and-improving-research/best-practice/nhs-site-set-up-in-england/frequently-asked-questions-about-research/) |
Guidance [first two columns] provided by the NIHR Clinical Research Network (now Research Delivery Network: RDN) (https://www.rdforum.nhs.uk/content/wp-content/uploads/2015/10/UTF-8CRN-principles-for-local-capacity-and-capa.pdf)
Arrange:
At this stage, the practice should put any necessary arrangements in place to deliver the study. Even when studies don’t require formal confirmation of C&C, there may be some arrangement required.
Please note that, if the study requires software to be added to the practice computers, this usually requires involvement from IT and you can seek advice from WY R&D.
The practice should consider each of the following arrangements, although these will vary by study:
|
Financial arrangements |
The financial arrangements applicable to the participating organisation are adequately described, i.e. the practice has full information/confirmation about which organisations will be paying for which costs. Any costs that the practice incurs should be covered by one of the following:
|
|
Internal arrangements |
Suitable arrangements for permissions/peripheral clinics/access when study involves NHS patients external to the participating organisation (e.g. another NHS Organisation) |
|
Emergency procedures |
Emergency procedures that may be necessary are conducted at the participating organisation in accordance with the protocol (e.g. to protect the participant in the event of a life-threatening incident or adverse event) |
|
Local contact information |
Where applicable the participant is clear about the local address and telephone number (usually included in the patient information), additionally contact details of the local investigator(s), and if applicable, other members of the research team, e.g. research nurses, Emergency contact information, if appropriate, contact information for complaints and, where appropriate, independent advisors. |
|
Archiving and storage arrangements |
Storage of data during the study and any post study archiving arrangements |
|
HRA guidance |
The HRA Approved protocol and any guidance provided in the HRA Approval letter are adhered to. This includes, but is not limited to, any patient consent, Information Governance, Human Tissue Act, and Mental Capacity Act arrangements. Radiation The Ionising Radiation (Medical Exposure) Regulations and the Medicines (Administration of Radioactive Substances) Regulations |
|
Fraud and Misconduct |
Local systems in place for dealing with fraud and misconduct |
Guidance provided by the NIHR Clinical Research Network (now Research Delivery Network: RDN) (https://www.rdforum.nhs.uk/content/wp-content/uploads/2015/10/UTF-8CRN-principles-for-local-capacity-and-capa.pdf)
Confirm:
At this stage, the practice confirms its participation (and confirms they are ready to start). The ways of doing this are varied depending on the study – this is confirmed in the HRA approval letter.
|
Involvement |
HRA letter wording |
What the practice must do |
|
PIC |
|
The HRA letter details the type of agreement the study is using. For a PIC, the HRA recommends that the model PIC agreement is used. This agreement is made between the practice and the site where the research activity is taking place. |
|
No formal confirmation required |
|
This is usually only for studies with minimal 'ask' for practices, or urgent public health studies where it is expected that all practices will participate. The research can proceed without agreement from the practice, after the length of time detailed in the HRA letter (in this example seven days). It is subject to the following conditions (as well as any others detailed in the letter):
If a practice does not wish to participate, they must give the sponsor a reason. If a practice positively confirms they can participate, then the study can proceed earlier than the stated length of time. Guidance can be read here: Where no C&C is required. |
|
Practices required to formally assess, arrange and confirm their participation |
|
The letter should also detail the method by which the practice will confirm C&C. Formal written confirmation must happen before any research activity can take place. This doesn’t mean wet ink signed contracts – in many cases a simple email will be acceptable as confirmation.
|



The next column in the table indicates the agreement to be used to confirm participation. See also the guide for contracts. Although the OID or the model agreement are the preferred contracts, you will need to use the contract which has been submitted and agreed by the HRA.
Most commonly, the standard agreements are used:
|
|
An authorised member of staff from the practice should check through the Localised Organisation Information Document (OID). This should detail what is required by the study, and all of the fields marked with a * should be completed by the study team, outlining the requirements. This should include start and finish dates, recruitment target, equipment and training requirements. The appendices detail any financial provisions, how materials will be shared and data processing/sharing arrangements. Items marked with a ^ are to be completed by the practice; then the OID should be returned by email from an authorised signatory (wet ink signatures are not expected). The Local OID is not a final document presented to the practice, but is to be negotiated and agreed between the practice and the study. Here is where you can agree targets, finances and local arrangements. |

An unmodified site agreement can also be used:
|
|
Unmodified agreements have been negotiated and reviewed by legal teams centrally, so practices can be assured that they can sign up to these, provided they have reviewed and agree to the study detail in the appendices. The PIC agreement is appropriate when acting as a Participant Identification Centre (PIC) [see Understanding the role of PICs guide]. It is an agreement between the practice and the research site (often a hospital trust). The person signing the agreement must be an appropriately delegated member of staff from the practice. An electronic signature will often be acceptable with agreement from the Sponsor. The signed copy should be kept in the site file (either hard copy or electronic) |
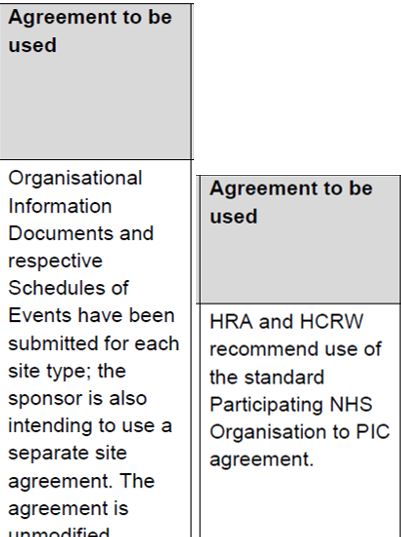
Modified/bespoke agreements are not recommended but can be used if agreed by the HRA:
|
|
It should be specified in the HRA letter where the Sponsor has used a modified or bespoke agreement. Where non-standard/modified agreements are used, the practice may wish to seek legal advice, but at the very least should carefully check through the agreement terms to make sure they are happy with these.
|
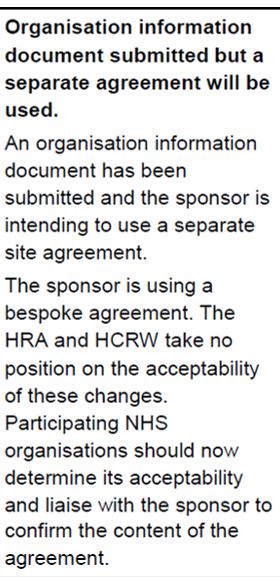
Sometimes, different parts of the study are subject to different agreements.
|
|
When different parts of the study are mentioned, the practice should confirm which parts of the study are applicable to them. The detail should be found in the OID, but if not sure you should clarify with the study team and receive confirmation in writing where possible. |
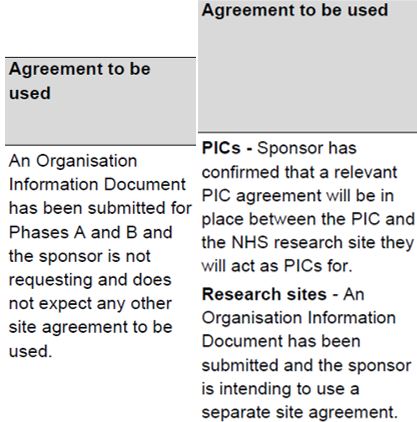
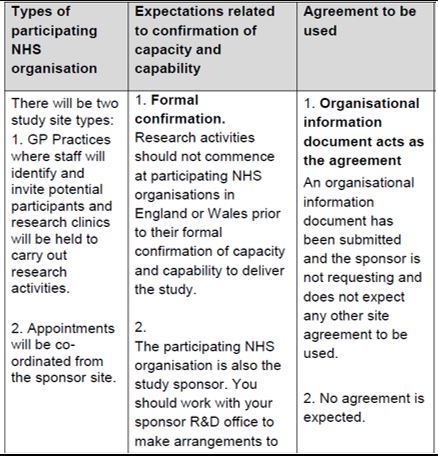
Other document types:
Studies which started a number of years ago may try to use older documents. The SSI (Site specific information) and Statement of Activities are now out of date so can no longer be used for confirming participation. The study team should be directed to https://www.myresearchproject.org.uk/help/hlpsitespecific.aspx#UK-Local-Information-Pack-Transition to replace these documents with the current ones.
Further advice and examples of setting up studies in primary care:
You can read more about study setup in primary care settings, including examples, at https://s3.eu-west-2.amazonaws.com/www.hra.nhs.uk/media/documents/HRA_Approval_in_Primary_Care_Settings__Principles_of_Study_Set-Up_Version_2.0_09Mar17.pdf (note that this document contains some old references but can be taken as the most recent guidance).
Glossary of Acronyms and Terms:
-
C&C - Capacity and Capability
-
RDN - Research Delivery Network
-
HRA - Health Research Authority
-
OID - Organisation Information Document
-
PI - Principal Investigator
-
PIC - Participant Identification Centre
-
SSI - Site Specific Information (form)
How to guide: Contracts
Some form of written agreement should be in place before a practice can start a study. Before agreement, the practice can discuss and plan for its involvement, but not recruit or start communicating with patients about the study.
Key information you should be aware of:
Agreements are put in place for any research or patient identification activity – this protects the practice as it clarifies what the practice is undertaking and makes sure all parties understand.
The Health Research Authority (HRA) strongly encourages the use of the Organisation Information Document (OID) or model agreements for agreement between studies and sites. When using a model agreement, the legal review has already been undertaken, so sites do not need to seek separate legal advice.
CTIMP or clinical device studies must have a written agreement – this is a legal requirement under clinical trials regulations.
Participant Identification Centre (PIC) sites should use the PIC agreement (model PIC agreement recommended). Studies can use other forms of agreement but practices may wish to seek legal advice if studies ask to use another agreement, as other agreements have not been verified by the HRA.
Note that a ‘signed’ agreement (even if not signed with a wet ink signature) is legally binding.
If practices are not sure of the requirements, they can contact West Yorkshire Research and Development (wyicb-bdc.research@nhs.net). WY R&D also offers advice to researchers, and can liaise with researchers about working with sites.
-
Signing up to be involved in a research study may involve signing a legally binding contract.
-
The HRA recommends that studies use model agreements which are standard templates having undergone review and confirmed as appropriate.
-
The agreement can only be signed by a person authorized by the practice to do this on the practice’s behalf.
-
Some studies will accept an email as confirmation of participation. Again, this should only be undertaken by an authorized person.
-
Some of these contracts take an ‘informal’ form but are still binding.
-
Studies may use other types of agreement but in these cases, practices may wish to seek legal advice.
-
The decision to participate, and the agreement, should be at the level of the participating organisation, i.e. usually at the level of the practice (as an independent contractor). PCNs are not able to sign a contract unless they have been set up as a company, and neither a PCN nor a federation is not able to sign a contract on behalf of its members unless there is a formal agreement that they can do this. If the research is taking place at the federation level, e.g. in the out of hours service, then this could be signed off by the federation. The ICB cannot sign off on behalf of practices.
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Principal Investigator guide
-
Data protection guide
Full guide
A contract will define roles and expectations, and in some cases is a legal requirement before a study can go ahead. It is helpful to ensure everyone has the same understanding of what is happening and who will carry out each activity, especially where transfers of data or tissue are involved, or financial arrangements.
Activity which can happen before a contract is signed:
Pre- agreement work can include:
-
Feeding into the planning and development of a study
-
Searching the practice database to see whether there are enough patients (to give an indicative number to the study team, but not contacting patients or sending any patient identifiable data to the study team)
-
Feasibility assessment to determine whether the practice can take part
-
Discussion with the study team and receiving documents to review the study
-
Signing of a non-disclosure agreement (sometimes required by commercial studies)
Studies not requiring a contract:
There will usually be no contract needed for studies which don’t require formal confirmation of capability and capacity. Practices are encouraged to confirm their participation via email to the study team. This would also enable the study to start as soon as the email (sent from an authorised member of staff from the practice) is sent.
Read more here: https://s3.eu-west-2.amazonaws.com/www.hra.nhs.uk/media/documents/collaborative-working-with-NHS-organisations-version-2-2-03-2018.pdf
If undertaking a study where the only activity is putting up a poster in the practice, then a contract will not normally be required.
Projects considered as service evaluation, rather than research, will not necessarily require a contract to be in place, but a data sharing or data processing agreement may be required if any data is involved. Contact us (wyicb-bdc.research@nhs.net) if you are in any doubt.
Who should sign?
The agreement should be signed by a person empowered to agree to the commencement of the research.
The practice should agree who will take on this role.
As the legal owners of the practice, the GP partners will usually be the people determining who is empowered to sign the agreement.
The ICB cannot sign any documents on behalf of the practice.
The decision to participate, and the agreement should be at the level of the participating organisation, i.e. usually at the level of the practice, unless the PCN or federation is the participating organisation, in which case the decision and agreement can be made by the PCN or federation. There must be a contractual relationship between the PCN or federation and the practice to allow the federation to conduct research on behalf of the practice. Contracts must be signed by an appropriate person with delegated authority by the practice.
What should happen before we sign?
Before signing, the practice should have completed an assessment of capacity and capability (C&C) to participate (see the guide above to assessing, arranging and confirming capacity and capability). The sponsor/study team should have sent all applicable documents to the practice, as well as sharing a copy with the local R&D team (West Yorkshire R&D). Practices should direct study teams to contact WY R&D (wyicb-bdc.research@nhs.net) and send the local documents, if not done already.
As your R&D department, the West Yorkshire R&D team will assess whether there are any potential issues in delivering the study, and will issue a C&C Advisory email to provide any advice we think will help practices in getting set up. WY R&D will send this to study teams, who can share this with practices to advise them. Practices can also contact WY R&D with any questions about the study or about their C&C assessment. See also the guide for assessing, arranging and confirming capability and capacity.
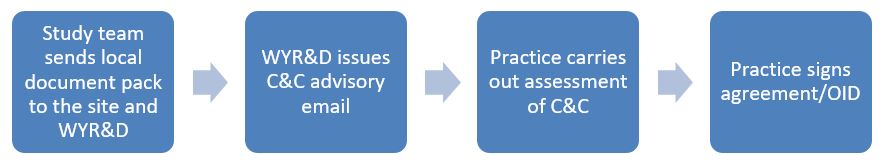
Types of agreement:
Below is a guide to the types of agreement usually used in research.
Organisation Information Document:
The Organisation Information Document (OID) is usually used to put in place arrangements between practices and the study (for non-commercial studies). It acts as a formal written agreement, including finances, transfer of biological samples, data sharing, data processing and intellectual property.
Appendix 1 of the OID confirms whether the sponsor intends to use the OID as the agreement with sites. Note that the OID becomes a legal contract between the practice and the sponsor. The appendices are part of the legal contract, if indicated within each appendix.

Signing of the OID counts as a practice’s confirmation of capacity and capability. The outline OID is the approved HRA document; following approval of the study the exact content of the OID is to be negotiated and agreed between the practice and the sponsor – for example the local PI, and the number of participants to be recruited. The OID is then revised as the ‘localised organisation information document’ and this is the one which should be signed.
If you would like to read more, the guidance for the OID can be found here: https://www.myresearchproject.org.uk/help/help%20documents/Guidance_Organisation_Information_Document_v1-8_April_2024.pdf
Signing the OID/contract:
It is not intended that wet ink signatures are used, so completing the authorisation section and returning the document by email (from an authorised person), counts as a signature.
Organisation information document for data processing:
If only undertaking data processing (for a non-commercial study), the recommended contract is a simplified Organisation Information Document (see this template).
Model agreements:
For Clinical Trials of Investigational Medicinal Products (CTIMPs) and Clinical Trials of Medical Devices, a written agreement is a legal requirement. The recommended agreement is the model agreement (unmodified).
Model agreements are available for research and for PIC activity. The HRA recommends that unmodified model agreements are used. You can look at the model agreements here.
Where other agreements or modified model agreements are used, practices may wish to seek advice.
PIC agreements:
A PIC agreement should be in place when the practice acts as a PIC for a study. See the guide on PICs for further information about being a PIC.
Use of an unmodified model PIC agreement is recommended. The agreement is put in place between the PIC and the research site – so for example a local Trust.
Other contracts:
If there are any Service Support Costs to be paid by the Research Delivery Network (RDN), practices will need to have a contract in place with the RDN (which puts in place payment arrangements). If you do not have a person to contact from the RDN, please contact wyicb-bdc.research@nhs.net and we will put you in touch.
Practices may want to consider implementing confidentiality agreements for researchers wanting to access their practice, as an extra reassurance. There is a template on the IRAS website: link
You may, usually for commercial studies, be asked to sign a non-disclosure agreement, which is used to ensure that commercially sensitive information is protected. This is common practice, but practices should ensure that they are happy with the terms and ensure that they can keep to the requirements. Be aware that these are legally binding so practices should be aware of their responsibilities under the contract and ensure they keep the confidential documents to the confidentiality standard required.
Glossary of Acronyms and Terms:
-
CRN - Clinical Research Network
-
GCP - Good Clinical Practice training
-
OID - Organisation Information Document
-
PI - Principal Investigator
-
PIC - Participant Identification Centre
-
SIV - Site Initiation Visit
-
SOPs - Standard Operating Procedures
-
CTIMP - Clinical Trial of an Investigational Medicinal Products
-
PCN - Primary Care Network
How to guide: Finance
It can be difficult to understand how studies are funded, what practices will be paid, and how. Funding can also come from multiple sources.
Key information you should be aware of:
-
Practices should make themselves aware of what finance is available and what they need to do to obtain this.
-
Practices may need to put contracts in place to enable funding to be paid.
-
Study teams should clearly outline payments.
-
Some payments may be variable depending on how many participants are recruited or on the number of mailout letters/texts sent.
-
Other funding may also be available to support practices.
-
NHS treatment costs or excess treatment costs are paid up front by practices but then paid back in full.
See also
-
Contracts guide
Full guide
Who pays?
Research guidance is clear on which organisation is responsible for paying for certain things.
You will be given information on these, but the below is provided for information to aid understanding about who is responsible for what.
|
Cost |
What it is |
Who is responsible for paying |
|
Research cost |
Costs of the R&D itself that end when the research ends. They relate to activities that are being undertaken to answer the research questions |
The study team (funded from the research grant). |
|
NHS treatment costs (also known as Excess Treatment Costs - ETC) |
Patient care costs, which would continue to be incurred if the patient care service in question continued to be provided after the R&D study had stopped, if these are higher than the cost of usual care. |
These are funded by the NHS and the Research Delivery Network (RDN) manages this. The funding itself comes from ICBs but is managed centrally. Studies do not automatically get these funded but need to apply. When there is an ETC incurred at the practice, the practice needs to pay out the cost first and it will be reimbursed. ETCs are reimbursed to practices at Q2 if they meet a minimum threshold of £100, or at Q4 if they do not reach the threshold. |
|
Service support costs (also known as study support costs) |
Additional patient care costs associated with the research, which would end once the R&D study in question had stopped, even if the patient care involved continued to be provided. |
The RDN pays these for non-commercial portfolio studies but again these are not paid automatically but need to be applied for by the study team.
|
Practices should:
-
obtain assurance that any costs incurred by the practice will be covered. Receiving an advisory email from the WY R&D team will include assurance of this.
-
ensure that the necessary contracts are in place to enable practices to get paid.
If you want to read more about this, please see: Attributing the costs of health and social care research & development (publishing.service.gov.uk).
Obtaining the finance:
The study team and RDN should provide information about how finance can be accessed relating to a study.
To obtain RDN funding, a practice needs a contract to be set up with the RDN. Contact yh.rrdn.studysupport@nihr.ac.uk if you have any questions about contracts. Service support costs are paid quarterly in arrears, so it may be some time after the activity when the practice actually gets paid.
To obtain research funding, the practice may need to invoice the study team. The study team should provide the detail of how this is done. The contract/PIC agreement/Organisation Information Document (appendix 2) should detail what will be paid and how. The practice may need to add its bank details to one of the appendices in the contract.
For PIC activity, the funding may be paid by the research site as this is the organisation who has the contract with the practices.
Other finances:
The RDN may occasionally offer additional funding to support practices with their research. There are regular funding opportunities and this will be updated when funding becomes available.
The RDN Agile Research Delivery Team also provides the opportunity for practices to request support to deliver specific studies – this can be applied for via https://sites.google.com/nihr.ac.uk/art/submit-a-request
Glossary of Acronyms and Terms:
-
RDN - Research Delivery Network
-
ICB - Integrated Care Board
-
ETC - Excess Treatment Costs
-
PIC - Participant Identification Centre
-
OID - Organisation Information Document
How to guide: Data protection
It is understandable to be nervous about data issues around research, including GDPR (now UK GDPR). Research can often require the sharing of information and it can be difficult to know what you can and cannot do. This guide attempts to summarise some of the key points but there is a lot of other guidance available (links below in Appendix 1) if you want to read more. You can also contact wyicb-bdc.research@nhs.net and we will be able to advise you.
Key information you should be aware of:
-
A practice is the data controller for its patient record system. However, data can also have a second controller (the research sponsor) when extracted for use in research. When data is created for a research project (e.g. a new measurement is taken or a research activity is recorded on a research document), then the sponsor is the data controller for this data.
-
Practices must be transparent and open with patients about how patient data is used and shared.
-
If you are undertaking research in your practice, you should mention this (and give detail) in your privacy notice.
-
In particular, when using patient data for sending information about research (e.g. contact details), this should be specifically mentioned in the practice privacy notice, to inform patients, ensure they are aware and there are no surprises, and give them the opportunity to opt out of receiving these communications.
-
You must support and respect the national data opt out. Practices have been required to comply since 31 July 2022. Automated services are available to make this easier to implement. There are cases where the opt-out does not apply, for example where a patient has consented to participate in a particular study. The opt out does not prevent patients being invited to participate in research, but their data cannot be used without consent.
-
You should only undertake research studies which have Health Research Authority (HRA) approval. This means any data sharing has been considered and agreed to be acceptable.
-
Studies must be conducted according to the approved study protocol, which should ensure compliance with applicable data protection and confidentiality laws.
-
Studies which are not ‘research’ but service evaluation or quality improvement, do not require HRA approval, so in these cases you may wish to seek advice (contact wyicb-bdc.research@nhs.net in the first instance).
-
You may have access to a Data Protection Officer (DPO) from the ICB and/or your federation who can provide advice.
-
It is worth noting that DPOs may not be familiar with research and the national approval processes. The HRA guidance will be the best place to consult for research-specific advice: https://www.hra.nhs.uk/planning-and-improving-research/policies-standards-legislation/data-protection-and-information-governance/gdpr-detailed-guidance/.
Full guide
This guide relates to research which has approval from the HRA. The HRA takes responsibility for reviewing and approving, amongst other things, the use and sharing of data. A practice can be assured that the data activities described in the approved study documentation have been confirmed as acceptable – so a researcher (or any members of the practice team) acting in accordance with the study protocol is approved by the national body.
In this guide, we address key points you should know and what actions you must take. We also give links to read more, and practices can always contact us to ask any questions. This guide also gives information about projects which are not managed as research.
Some useful training can be found here: https://byglearning.com/mrcrsc-lms/course/view.php?id=71 (an account is required but can be created via this link for free).
Data controller:
Although the practice is the data controller of the patient record system, if data is extracted for or by a research study or created in the process of the research, then the research sponsor of the study is the data controller of this data for the purposes of research. If data is created by a member of the practice team (e.g. for completing a case report form (CRF)) for the purposes of research, then the research sponsor is the data controller and the person completing the form is the data processor.
Privacy notice:
If you are undertaking research in your practice you should mention this in your privacy notice, to be displayed on your practice website as well as in your practice, particularly if you produce information for new patients joining the practice. The law says that you must be transparent about data that you collect from patients. You should explain what you are collecting and sharing, why and how, and how patients can object (it is not sufficient to just say ‘research’ – e.g. the practice team may look at your record to determine whether you are suitable to be invited for a research study.) The notice should explain enough that the patient can understand what is being done and there should be no surprises. If someone other than practice staff will have access to any records, then this should be mentioned in the privacy notice.
If your practice works with any third parties and shares data with these for the purposes of research, then patients should be informed about this and given the opportunity to opt out.
The practice should think about means of communicating the privacy notice to patients – for example a text message campaign or recorded message on the phone system, which could direct patients to read the notice on the practice website.
The BMA produces templates and posters you can use, so you can be confident that these meet legal requirements: https://www.bma.org.uk/advice-and-support/ethics/confidentiality-and-health-records/gdpr-privacy-notices-for-gp-practices.
You can read further advice about text messaging to patients at https://www.themdu.com/guidance-and-advice/guides/text-messages-in-general-practice
Action: Make sure research is mentioned in your privacy notice and displayed/communicated to patients. You should clearly explain what will be done with a patient record.
National data opt-out:
The national data opt-out gives patients the opportunity to opt out of the use of confidential patient information for use in planning and research. That means that if you share data for research, the practice is responsible for ensuring that people who have opted out are removed from that data. You must exclude patients who have indicated an opt-out. The data opt-out does not apply where the patient has individually consented to take part in a particular study (and in some other circumstances). Patients who have opted out of their data being shared for research can still be invited to participate in studies by the practice team, however you must not disclose patient data to a third party (e.g. research team) without excluding patients who have opted out.
The national data opt-out is now mandatory (from 31 July 2022).
The national data opt-out is now the primary means of patients opting out of data sharing for research and planning. The previous type-1 opt-outs (recorded at the practice) have now been converted to the national data opt-out, although patients can still record these. This will probably be discontinued as the system moves to the national data opt-out.
Parents/guardians can also make the same choice on behalf of a child under 13. Children 13 or over can opt out for themselves.
Opt-outs do not apply in the following cases:
-
For a patient’s direct care;
-
Where a patient has given their consent to the specific research and to the sharing of data [this does not extend beyond what they have consented to in the specific study];
-
In a small number of cases using section 251 approval (see below);
-
For data which has been fully anonymised under ICO guidance (see section on anonymisation below);
-
For aggregated data.
Opt-outs do apply where:
-
Data is being shared for purposes outside of direct care and none of the above applies;
-
Section 251/CAG approval is in place (see section below).
Opt-outs continue to apply after death, but do not apply retrospectively, so will not need to be applied for data that has been previously shared.
Action: Practices must:
-
Put in place a system for applying data opt-outs. Even if you do not currently have any data which requires checking of the opt-out, you need to be ready to implement a solution in case a situation occurs for which the opt-out check is needed.
Practices are recommended to:
-
Register for MESH , a system which allows NHS numbers in any data request to be checked against national data opt-outs held in the SPINE: https://digital.nhs.uk/services/message-exchange-for-social-care-and-health-mesh
Implementing the data opt out:
-
Every time you are asked to share confidential patient information, you will need to use a service like MESH to compare NHS numbers with the opt-out list. When you send a list via MESH to the National Data Opt-out Service, you will receive back a list with the opt-outs removed.
-
You must remove the entire record of any patient who has an opt-out from any data you share or use for research or planning purposes.
-
Practices must check all requests against the opt-out list.
-
Researchers asking to view a patient record with consent, should share with the practice a copy of the consent form or other evidence of consent (consent overrides any opt-out).
-
If you re-run a search for any reason (e.g. follow up mail-outs) you may need to re-check against the opt-out list.
You can read more about the technical solution for implementing the opt-out online: link
See https://digital.nhs.uk/services/national-data-opt-out/compliance-with-the-national-data-opt-out for details on complying with the opt-out.
The following training from e-Learning for Healthcare is useful if you have any further concerns. You need to register for an e-LFH account if you don’t already have one: https://www.e-lfh.org.uk/programmes/national-data-opt-out-training/
Participating in a research study;
Your practice may be invited to be involved in research studies and should bear in mind the following:
-
Studies are reviewed and approved by the Health Research Authority (HRA) and Research Ethics Committee (REC) where applicable. If you receive a study you should ask to see the HRA approval letter and an approved study protocol which will detail all activities.
-
The HRA considers all issues relating to data, including data sharing.
-
The HRA recommends that studies use template agreements, including data sharing agreements. Practices can be confident in the use of template agreements as these agreements have been reviewed by legal representatives and approved.
-
Studies must be conducted in accordance with the study protocol (this is an HRA-approved document) and practices and study team who are working in line with an approved protocol can be assumed to be acting in accordance with all legislation.
-
If a study does not have a letter of HRA approval, practices should contact the WY R&D team (wyicb-bdc.research@nhs.net) for advice.
Access to patient data:
If any researchers ask to access your practice or practice record system, it is the practice’s decision whether to allow this. You should bear in mind the following:
-
A researcher should have a Letter of Access (see staff guide), issued by the WY R&D team at the ICB. This provides assurance that the researcher has appropriate qualifications and approvals for accessing data.
-
A researcher should only have access to the data which allows them to carry out the research, so should be restricted.
-
A researcher may need a smart card to access the system, which can usually allow their access to be restricted.
-
Researchers may not use another practice staff member’s smart card.
-
When allowing access, you should ensure that any researcher is aware of, and willing to comply with, NHS Information Governance requirements. You can print and give them this guidance for reassurance.
-
Contact https://digital.nhs.uk/services/registration-authorities-and-smartcards/care-identity-service for advice on smart cards.
-
Researchers should return smart cards after the research project finishes, and/or their access to patient records should be terminated.
-
Information on allowing access to electronic patient records for researchers can be found here: https://www.gov.uk/guidance/on-site-access-to-electronic-health-records-by-sponsor-representatives-in-clinical-trials
Action: Ask to see all researchers’ letters of access if accessing patients or data.
Patient Identifiable Data before/without consent:
In most cases, where studies involve accessing patient identifiable data before consent (e.g. for the purposes of inviting a patient to participate in a study), then this may only be accessed by someone who is a member of a patient’s direct care team.
‘Member of the direct care team’ usually means a member of staff at the practice, or in some cases where there are groups of practices, it could be a member of staff at another practice in the Primary Care Network (PCN), or someone employed by the federation, if they can meet the following criteria:
-
A person who has a legitimate relationship with the patient;
-
Someone who has access to the patient’s record as part of their existing role.
In these cases, there should be a formal employment contract or data sharing arrangement between the person and their employing organisation, and this access should be mentioned in the practice's privacy notice.
A study’s plans for any access to patient data before consent should be outlined in the study protocol, and this has been reviewed and approved by an ethics committee and the HRA – so by following the study protocol practices can be assured that the way of working is approved. In case of any concerns, contact WY R&D. If a study team requests to access data using methods not outlined in the study protocol, then you can also contact WY R&D for advice.
Projects which aren’t managed as research:
Projects may (under specific conditions) be managed as service evaluation, audit or quality improvement, which may mean they do not require formal research approvals.
-
If a project is only assessing existing care, rather than introducing a new treatment, or testing a hypothesis, then it can class as evaluation or audit.
-
If a project randomises participants, then it must be managed as a research project and apply for HRA approval.
-
If you would like reassurance that a project is not research, you can direct the project leaders to contact WY R&D (wyicb-bdc.research@nhs.net) and we will assess the project and can issue a letter confirming this. Any questions you may have feel free to contact us or direct the team to contact us.
Freedom of information (FOI) legislation:
ICB FOI requests can be made here: https://www.westyorkshire.icb.nhs.uk/contact/submit-information-request. Research information may be exempt from Freedom of Information legislation, if the release of the information may prejudice the continuing research and/or publication. If you receive a freedom of information request, contact the ICB governance lead for further advice.
Appendix 1
Further information to help with queries or concerns about data for research
You may get queries from patients or staff about participating in research studies, so below is some further information. It is not expected that practices need to be familiar with the below, but practices may have concerns and queries, so this information is provided for reassurance. In addition, the HRA, WY R&D and NIHR Research Delivery Network (RDN) are all available for any further queries. Your practice may also have access to advice from a DPO (usually provided by the federation or ICB) – please bear in mind that research is a specific case in terms of data protection law, so DPOs may not be familiar with all of the detail in research and you may wish to seek advice from wyicb-bdc.research@nhs.net.
Section 251 (also known as Confidentiality Advisory Group or CAG approval):
-
Section 251/CAG approval may apply to research projects which need to access patient identifiable data without or before consent. Researchers have to apply for this approval alongside their other applications.
-
Most research projects cannot get this approval, if the Confidentiality Advisory Group considers that they could access the data in another way. Section 251 is there for the purposes of enabling research which cannot feasibly be achieved if there was a requirement to obtain consent from every participant.
-
A section 251 exemption does not require a practice to process/share the data, but gives a legal basis to enable your practice to participate in the research.
-
You should ask to see a copy of the CAG approval letter.
-
This is something that the research study would undertake, rather than something the practice needs to do. It will form part of study approvals and usually would need to be put in place at the planning stage.
Anonymised/de-identified or pseudonymised data:
It is best practice to minimise the sharing of personal confidential data and this will mean using anonymised data where possible or reducing the number of identifiers shared.
Data which is truly anonymised is not subject to GDPR, but care should be taken when referring to data as ‘anonymised’ as it is often not fully anonymous and can be reidentified with either existing knowledge or if in conjunction with other information. The term ‘de-identified’ is sometimes used to denote data which has had identifiers removed.
If asked to produce de-identified data, practices must:
-
Be confident that the risk of being able to reidentify any participant is very small (note the risk is never zero but should be as low as possible)
-
Consider suppressing numbers of 5 or less
Anonymisation/de-identification should happen in the practice and be carried out by someone legitimately allowed to view/process the data (usually a member of the direct care team) before it is sent to an external researcher.
-
Data are often pseudonymised – which applies to any data which has a pseudonym applied to allow data to be matched up with other sources. The NHS number is an example of a pseudonym, or a study ID number.
-
Pseudonymised data does not class as anonymised under ICO guidance.
-
Often it is necessary for a study team to be able to reidentify participants, for example when if there is a safety concern, there needs to be a way for the research team to identify which treatment(s) a patient has received. Or if a patient requests to be withdrawn from a study, their record will need to be identified in order to remove it.
-
If someone has access to the pseudonymisation ‘key’, or other additional information (for example postcode), they could feasibly identify the person. Access to the pseudonymisation key must be closely restricted – this should be detailed in the protocol.
-
A practice should be assured that the security measures used will reduce the possibility for re-identification.
COPI notice:
The Control of Patient Information (COPI) notice, which allowed organisations to share patient data to support the COVID-19 response, including research, has now expired. Any research which was using this notice as its legal basis for sharing, must now have an alternative legal basis for this. Practices must ensure that any data they share has a legal basis for doing so. If they are not sure they should query this with the study team, the ICB Data Protection Officer, or the ICB research team: wyicb-bdc.research@nhs.net.
A new notice has been issued under COPI regarding COVID-19 information through the OpenSAFELY data analytics platform: https://www.gov.uk/government/publications/covid-19-notification-to-gps-and-nhs-england-to-share-information.
Sharing data between the UK and other countries:
There may be cases where personal data is shared with other countries as part of a research project – for example storing data in another country including perhaps using servers located in another country. This is permitted with other EU countries, and also Andorra, Faroe Islands, Guernsey, Isle of Man, Israel, Japan, Jersey, New Zealand, Switzerland and Uruguay. If the project is sharing data with any other countries, you may wish to read more about this.
There is a more problematic issue, if your practice is involved in any research projects which involve receiving data from the EU (or other countries). This is unlikely as most of the time practices are involved with the use of their own data.
The EU and UK have come to a ‘data adequacy decision’ which allows the free flow of data between the UK and EU. This means that the EU has agreed that the UK data protection safeguards are enough to allow data to flow between the EU and UK the same as between EU member states.
Any project which includes sharing of data outside of the UK should have included this in their HRA and REC submission, to be reviewed by these bodies. If you share data in the ways outlined in the study protocol, this is approved and indemnified as part of the HRA approval. In other words, you are not responsible for checking and understanding the detail of data sharing, but as long as you act in accordance with the study protocol, then you can be assured that this has been approved by the responsible body.
You can read more at:
Glossary of Acronyms and Terms:
-
RDN - Research Delivery Network
-
ETC - Excess Treatment Costs
-
PIC - Participant Identification Centre
-
OID - Organisation Information Document
-
GDPR - General Data Protection Regulation
-
DPO - Data Protection Officer
-
ICO - Information Commissioner's Office
-
COPI - Control of Patient Information
-
LoA - Letter of Access
-
PCN - Primary Care Network
How to guide: Following protocols
A protocol is the description of everything which will take place within a research project.
-
The protocol is the instruction manual for a research project.
-
It should contain all of the things that you need to do.
-
It is the reference guide for all activities which have been approved to take place.
-
All study staff should be following the latest approved version of the protocol.
-
You should not undertake any activity which is not in the protocol.
-
It should be kept up to date; when there are any changes, a new version of the protocol should be issued and an amendment submitted to approve the changes. Changes should not be implemented until a new version of the protocol has been approved by the HRA. Staff delivering the study must not make changes - these must be done by the study team centrally.
Full guide
Format:
-
The protocol is not always in a standard format – although the Health Research Authority (HRA) does recommend a template for CTIMPs and qualitative research: https://www.hra.nhs.uk/planning-and-improving-research/research-planning/protocol/
-
The protocol may be in printed and/or electronic format.
Keeping records:
-
The protocol should be kept in the site file.
-
Superseded versions should be kept in the file but crossed through (or marked as superseded if kept electronically).
-
Staff involved in the study must have access to the site file to enable them to refer to the protocol.
Glossary of Acronyms and Terms:
-
HRA - Health Research Authority
-
CTIMP - Clinical Trial of an Investigational Medicinal Product
How to guide: Receiving consent
This guide outlines some general guidance on receiving consent.
Key information you should be aware of:
Informed consent is a fundamental principle of research and it is vital that consent is given freely, with full information of what a participant is consenting to being provided. Either the participant must have capacity to consent or it must be received by means of a process involving a consultee or legal representative (where this is appropriate and allowed for in the study). Consent should be received only by a member of staff who is appropriately trained and approved to do so. Consent may be withdrawn and withdrawal should be respected wherever possible.
-
The practice is responsible for ensuring that all staff who may be involved in a study have an understanding of what is involved.
-
Staff involved in the consenting of patients may need to complete Good Clinical Practice (GCP) training. This is a legal requirement for Clinical Trials of Investigational Medicinal Products (CTIMPs) and may be a requirement specified by the study protocol.
-
All staff involved in research at the practice must have completed study specific training where this is required for the study.
-
Consent must have been received before any research activity relating to the patient and/or their personal data takes place.
-
The consent process must be conducted in line with GCP principles and the study protocol.
-
Consent is not always written, and there may be instances where researchers can use verbal consent, online consent, or other proportionate methods to reduce burden for both the participant and the study.
-
There could be instances where it is not possible for participants to be completely withdrawn, e.g. if the data or samples have already been processed, anonymised and aggregated, but this should be fully explained within the consent process as well as the study protocol.
Highlights
-
Preparation
-
Consent process
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Site selection / initiation visit (SiV) guide
Full guide
Preparation:
The Health Research Authority (HRA) approvals process includes ensuring that the consent process for approved research is ethical and legal. Therefore, it is vital that the consenting of participants is carried out in line with what is stated in the current HRA approved study protocol.
Staff involved in the consenting of participants must be:
-
adequately qualified as outlined in the study protocol or by the study team (this may be specific to particular studies i.e. some studies may require that the consenting of participants must be done by a nurse or GP);
-
approved to do so by the Principal Investigator of the local site (GP practice) and this must be included in the Delegation Log.
For CTIMP studies or if specified in the protocol, staff receiving consent should be GCP trained and have a certificate that is recent enough for the requirements of the study (usually within 2 years – if in doubt, this should be checked with the study team);
Consent process:
Participants must be fully informed and in a position to consent (having capacity to consent or by means of a process involving a consultee or legal representative where this is appropriate and allowed for in the study). See GCP or Informed consent training for further information.
Examples where someone may not be able to consent for themselves are children, those with diminished capacity or people who are unconscious.
The term 'receive consent' is preferred rather than 'take consent'.
The HRA is encouraging a proportionate approach to consent for low risk studies https://www.hra.nhs.uk/about-us/news-updates/hra-publishes-new-proportionate-consent-guidance/
Consent must have been received before any research activity relating to the patient and/or their personal data takes place.
Consent must be documented and filed in line with the study protocol.
Since consent is an ongoing process, staff involved with study participants should ensure they are happy to continue during study follow-up visits etc. and must respect any wishes to withdraw consent.
Specific training is available via NIHR Learn on consent for children, adults lacking capacity and for different communities (https://learn.nihr.ac.uk)
Further general guidance on the subject of consent in research can be found online: http://www.gov.uk/service-manual/user-research/getting-users-consent-for-research#what-informed-consent-is
Glossary of Acronyms and Terms:
-
HRA - Health Research Authority
-
GCP - Good Clinical Practice
How to guide: Managing research staff arrangements
This guide outlines the responsibilities of practices in managing external research staff and internal staff working on a research project.
Key information you should be aware of:
-
Please note that external research staff could be from another NHS organisation, from a Higher Education Institute (HEI), from a Health and Social Care organisation (HSC) or be a staff member of the National Institute for Health Research (NIHR) Research Delivery Network (RDN).
-
The practice Principal Investigator (PI) is responsible for ensuring that any external research staff have been appropriately screened and have appropriate training and experience.
-
Practices should ask to see an external research staff member’s letter of access (LoA) before granting them access to the practice to support a study. Letters of Access are provided by WY R&D to verify the above.
-
WY R&D provide this service to all West Yorkshire general practices using the NIHR Human Resources (HR) Good Practice Resource Pack.
-
Internal staff must be suitably qualified and trained to undertake research activities; this does not require additional LoA, but the Principal Investigator must be confident of their abilities and must delegate duties to staff using the delegation log.
-
The practice PI is responsible for delegating research activities via the study delegation log to all research staff working on a study, whether internal or external.
Highlights
-
Processes
-
Practice and principal investigator (PI) responsibilities
Also see
-
Roles and terminology guide
-
Principal Investigator guide
-
Delegation log guide
Full guide
The HR Good Practice Resource Pack has been reviewed and updated in light of the Data Protection Act 2018 (DPA 2018) and the UK General Data Protection Regulation (GDPR) which came into force in the UK on 25th May 2018.
Processes:
-
As recommended by the Department of Health and Social Care (DHSC) the West Yorkshire R&D team uses the HR Good Practice Resource Pack to support the West Yorkshire general practices by issuing letters of access (LOA) where appropriate.
-
The HR Good Practice Resource Pack contains information and documentation to support the process for handling HR arrangements for researchers and provides a streamlined approach for confirming details of the pre-engagement checks they have undergone with the NHS/HSC.
-
It applies to NHS/HSC organisations and HEIs in England, Scotland, Wales and Northern Ireland, and has been developed in parallel with other arrangements across the UK to streamline the process for setting up research in NHS/HSC organisations.
The Resource Pack includes details of:
-
A Research Passport system for issuing honorary research contracts (HRCs) or letters of access to HEI researchers who need to undertake their research within the NHS/HSC. The research passport provides evidence of the pre-engagement checks undertaken on the researcher in line with NHS Employment Check Standards; and
-
Arrangements for sharing and accepting pre-engagement checks between NHS/HSC organisations when NHS/HSC staff wish to undertake research within the NHS/HSC outside of their employing organisation.
The WY R&D team cannot issue honorary contracts.
Link: HR Good Practice Resource Pack
Practice and principal investigator (PI) responsibilities:
The practice PI is responsible for ensuring that any external research staff have been appropriately screened and have appropriate training and experience. Reviewing the external research staff member’s letter of access is sufficient to ensure this for studies that are not Clinical Trials of Investigational Medicinal Products (CTIMPs).
-
PIs should check the letter is in date and references the correct study. For CRN staff this may be a blanket letter for all NIHR studies.
-
It is considered best practice to keep a copy of the letter of access in the site file.
The practice PI is responsible for delegating research activities via the study delegation log to external research staff:
-
Delegated research activities to external research staff must be POST participant consent unless an appropriate agreement (NOT JUST THE LoA) is in place to make the external research staff member a member of the direct care team.
-
For CTIMPs it is considered best practice to keep a copy of the research staff member’s CV and appropriate training for research activities on the site file. The PI should request these from external staff before delegating these research activities.
-
Should the practice PI wish to hold a copy of the external research staff member’s CV and training certificates for a non-CTIMP study, they may request these from the external research staff member before delegating any research activities.
Glossary of Acronyms and Terms:
-
CRN - Clinical Research Network
-
CTIMPs - Clinical Trials of Investigational Medicinal Products
-
DHSC - Department of Health and Social Care
-
GCP - Good Clinical Practice training
-
HR - Human Resources
-
HSC - Health and Social Care (the equivalent of the NHS in Northern Ireland)
-
LoA - Letter of Access
-
PI - Principal Investigator
-
HRC - Honorary Research Contract
How to guide: Amendments [NOTE THAT NEW REGULATIONS ARE REPLACING THE TERM "AMENDMENTS" WITH "MODIFICATIONS"]
When a study starts, the protocol and documents are all approved by the Health Research Authority (HRA) and research ethics committee (REC). If there are any changes to documents, processes or dates, these usually have to be notified to the HRA/REC and be reviewed again. A separate approval of this is then issued.
Key information you should be aware of:
-
The practice is responsible for ongoing treatment for its patients, and for ensuring that patients and patient data are treated with due care.
-
The practice must ensure that it is always using the current approved documents when running a study – this is an essential part of following a protocol.
-
The study team is responsible for updating the study and submitting any amendments to the HRA/REC for approval. The practice team cannot make amendments to the study.
-
Urgent safety measures can be implemented without amendment approval; in these cases the sponsor will inform you of the change and inform you that it is an urgent safety measure.
-
The study team is responsible for letting practices know about all amendments and for sharing documents as appropriate. They must let all sites know of any changes to the study which affect them.
-
Practices do not need to send a formal acceptance of an amendment, and the sponsor may implement the amendment if the practice does not object or request more time to consider the amendment, providing the amendment is approved and the appropriate time has elapsed.
-
Amendments are named by the sponsor and there is not a defined numerical system, which can lead to confusion; amendments may not seem to run chronologically. You should be informed about all amendments which apply to your practice and it is the sponsor’s responsibility to keep you informed.
-
If a practice feels unable to implement an amendment, they must communicate with the sponsor as they may not be able to continue with the study without the amendment.
Highlights
-
Assessment of feasibility
-
Submitting EoIs
See also
-
Getting your practice ready to do research guide
-
Roles and terminology guide
-
Principal Investigator guide
-
Data guide
Full guide
Amendments are a necessary part of studies, as they allow the study to respond to emerging requirements and correct any errors.
Amendment processes:
-
When a change needs to be made to a study (which can be as small as an alteration to the text of one study document), an amendment is required.
-
The study team identifies a need for an amendment.
-
If practice staff identify a need for an amendment, they must communicate it to the sponsor as the sponsor is responsible for making any changes. They must not make changes without the sponsor’s agreement – amendments must go through the process.
-
The sponsor submits the amendment, and the automated tool (the amendment tool) determines whether it needs review by the HRA. The amendment may also need to be reviewed by a research ethics committee.
-
The amendment tool will produce a decision so the study team can share this with sites.
-
The changes may not be made until the amendment has been approved, or the amendment tool has confirmed that approval is not required. You should receive copies of the amendment tool and any approvals.
-
The study team needs to share information and updated documents with sites. They may use the following email templates:
Template email for sponsors to share category A or B amendment documents with sites (regulatory approvals outstanding)
Template email for Category A or B amendment documents with sites – where regulatory approvals in place
Template email for sponsors to share category C amendment documents with sites
Template email for sponsors to confirm implementation of an amendment
-
The study team also needs to share details of the amendment (plus approval where applicable) with West Yorkshire Research & Development (WY R&D) as your NHS R&D team, and WY R&D will produce an advisory email for the study team, which they are then asked to share with practices.
-
The site (practice) must update the site file and documents being used (see section below on updating files).
-
The study team may use an amendment log (e.g. https://www.myresearchproject.org.uk/help/hlpamendments.aspx#AmendmentLog) to record and keep track of amendments; they may also provide this alongside amendment notifications to help participating sites to keep track as well.
Amendment categories:
Amendments are categorised to determine whether the site (practice) needs to review them before implementing them.

Urgent safety measures:
Where patient safety is at risk, urgent changes may be implemented but these must be driven by the sponsor. If you feel the study is creating a risk, you must report it to the sponsor rather than making any changes.
The sponsor must inform all sites of the change. There will often be changes to documents following an urgent safety change – these should be submitted as usual as an amendment.
Practice responsibilities:
The practice is responsible for delivering the study in accordance with the study protocol, including all amendments.
The study team is responsible for making any updates to the study documents, and securing all necessary approvals for these. The practice is responsible for replacing all documents with the latest versions in the site file, and only using the current document versions.
The Principal Investigator (PI) must ensure that all staff members are informed of any changes. The delegation log may need to be updated if staff take on new activities.
The sponsor will determine whether participants need to receive updated versions of documents. When the consent form is updated, the sponsor will determine whether participants who have already given consent, need to then sign a new consent form.
Updating files and paperwork:
A site file must contain all study documents.
When a document is replaced with an updated version, the following process must be followed:
-
Retain all old versions in the study site file.
-
If paper versions, the old version should be crossed through and marked ‘superseded’ (the old versions should be retained in the file clearly marked) and the new version inserted.
-
If using electronic site files, it must be made clear which are the current and which the archive versions, for example saving the previous versions of documents in a separate ‘archive’ folder.
Amendment approvals should also be included in the site file.
Agreeing to or rejecting an amendment:
Practices do not need to send a formal acceptance of an amendment, and the sponsor may implement the amendment (when approved) if the practice does not object or request more time to consider the amendment. They may implement category A or B amendments 35 days after they notify the site and may implement category C amendments as soon as they have notified the site. If the practice confirms to the sponsor that they accept the amendment, then it can be implemented sooner than 35 days.
Note that days means calendar days and not working days.
If your practice feels it cannot implement an amendment, it must raise an objection in writing to the sponsor as soon as possible. This must be within 35 days.
If your practice needs more time to consider an amendment, it must notify the sponsor (again, this must be within 35 days). However, it is best practice to progress amendments (or object) as quickly as possible.
Amendment naming and numbering:
There is no standard way of naming and numbering amendments. Sometimes the terms ‘minor’ and ‘major’ are used, sometimes ‘substantial’ and ‘non-substantial’.
The definitions ‘minor’, ‘major’, ‘substantial’ and ‘non-substantial’ are simple descriptors – they all still need to be implemented.
Categories of amendment (A, B and C) are more relevant (see above) as they describe whether the amendment needs management or oversight from the site (practice).
Some studies prefer to number amendments starting at 1 and numbering each amendment chronologically; others number substantial amendments separately from non-substantial amendments. Others may use the date or a description rather than a number. See https://www.myresearchproject.org.uk/help/hlpamendments.aspx#AmendmentLog for information an amendment log which sponsors may choose to use to keep track of and record amendments.
A practice may not always receive every amendment, as category B amendments are not necessarily relevant to all sites (for example if a new site is added to a study, or if a change is made to a different arm of the study). It is the sponsor’s responsibility to inform the practice of all amendments applicable to them.
Changes not requiring an amendment:
If a study document contains a section designed to include local information or branding (see example below), or information that is not known at the time of the approval (for example a specific URL), then it does not require an amendment, if this is clear at the point of application.

Changes to an Organisation Information Document are a matter between the site and sponsor, so do not require an amendment.
When the study was approved, it will have specified a start and end date. If there are any changes to the study end date, particularly if they are substantial, they will usually be submitted as amendments.
In exceptional circumstances, there may be exemptions to requiring a study amendment. In recent years, the most important to be aware of are:
-
For studies affected by the COVID-19 pandemic, for example those which had to pause or implement new methods, restarting or reverting to original methods does not require an amendment, but must be discussed with sites.
-
With the introduction of the General Data Protection Regulation (GDPR), the HRA produced template wording to be used in research documentation. If studies used the template wording, they did not require an amendment, but simply needed to notify sites of the update.
If you are not sure, then please contact wyicb-bdc.research@nhs.net or discuss with the study sponsor.
Glossary of Acronyms and Terms:
-
RDN - Research Delivery Network
-
GCP - Good Clinical Practice (training)
-
OID - Organisation Information Document
-
PI - Principal Investigator
-
SIV - Site Initiation Visit
-
SOPs - Standard Operating Procedures
-
HRA - Health Research Authority
-
REC - Research Ethics Committee
How to guide: Study conclusion (closedown)
This guide outlines general guidance on the process for study conclusion (closedown).
Key information you should be aware of:
-
The practice is responsible for the ongoing treatment of its patients, and for ensuring that patients and patient data are treated with due care.
-
Sponsors are responsible for the management of a specific study.
-
Chief Investigators (CIs) are responsible for the conduct of a specific study within the UK.
-
Archiving of study documentation is necessary to enable any subsequent evaluation of study conduct.
-
The Clinical Trials Regulations and specifically, Regulation 31A of the Medicines for Human Use (Clinical Trials) Amendment Regulations 2006, define the archiving requirements for Clinical Trials of Investigational Medicinal Products (CTIMPs). See the NIHR Toolkit for Archiving: https://www.ct-toolkit.ac.uk/routemap/archiving/
Highlights
-
Study conclusion
-
Archiving
See also
-
Roles and terminology guide
-
Data protection guide
-
Retention of documents guide
Full guide
Study conclusion:
-
The protocol or subsequent agreement(s) will determine the date for the conclusion of the study.
-
The study team should inform the local Principal Investigator (PI) of the closure of the study.
-
Some studies may conclude before the conclusion date or event for a variety of reasons.
-
The site (GP practice) should keep a record of study conclusion dates.
Archiving:
-
Archiving of study documentation is necessary to enable any subsequent evaluation of study conduct.
-
The study protocol should outline the process for archiving study documents including the length of time these need to be kept. If there is any doubt, please contact the study team.
-
Archiving should be carried out in line with the study protocol alongside your local policy for archiving and storage.
Glossary of Acronyms and Terms:
-
CI - Chief Investigator
-
PI - Principal Investigator
How to guide: Retention of documents
Records management relates to individual, professional, and organisational responsibilities to manage and retain records effectively and consistently.
Key information you should be aware of:
-
In England, all NHS organisations and managers need to enable staff to meet the standards set out in the Records Management Code of Practice: Records Management Code of Practice
-
Practices should ensure good practice is adhered to in relation to records management, as per national guidance.
-
The practice Principal Investigator (PI) should adhere to the records management and retention requirements that are outlined in research study protocols. This activity can be delegated via the study delegation log.
Highlights
-
The Records Management Code of Practice provides guidance for those working within, or under contract to, the NHS in England.
-
Standards and practice will change over time, so the Records Management Code of Practice document will be reviewed and updated when needed. Practices should ensure the most recent version of this records management guidance is followed by visiting the NHS England website: Records Management Code of Practice
-
The code has a section dedicated to the required retention periods for clinical trials and research. The retention schedule can be found in Appendix II.
-
When supporting specific studies, practices should consult the study protocol which will outline the site data retention and management requirements.
See also
-
Study conclusion (closedown)
Full guide
Guidance is available to advise of good practice in relation to records management within the NHS in England. Practices should ensure they uphold high standards of record management by following national guidance. The practice PI should follow the records management and retention requirements that are outlined in research study protocols, reviewing these for each individual study. This activity can be delegated via the study delegation log.
Records management and retention:
-
The Records Management Code of Practice is a guide for NHS organisations in England to follow when managing records: Records Management Code of Practice
-
This provides a framework to support consistent and responsible records management. This code considers individual, professional, organisational, and legal responsibilities when managing records.
-
Practices should refer to the NHS England website for the most current version of this guidance. The code has a section dedicated to the required retention periods for clinical trial and research records.
-
Practices should ensure good practice is followed when managing records, in liaison with study teams. When supporting specific studies, practices should consult the study protocol which will outline study specific data retention and management requirements. Practices should contact the study team to discuss if further information is required.
Glossary of Acronyms and Terms:
-
PI - Principal Investigator